- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Pyocyanin Extraction and Quantitative Analysis in Swarming Pseudomonas aeruginosa



Published: Vol 6, Iss 23, Dec 5, 2016 DOI: 10.21769/BioProtoc.2042 Views: 16809
Reviewed by: Valentine V TrotterAmit DeyAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Jan 2016
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
This protocol describes the quantification of pyocyanin extracted from swarming colonies of Pseudomonas aeruginosa. Pyocyanin is a secondary metabolite and a major virulence factor, whose production is inducible and varies highly under different growth conditions. The protocol is based on the earlier developed chloroform/HCl extraction of pyocyanin from liquid cultures (Frank and Demoss, 1959). Swarming colonies together with the agar they occupy are split into two halves. Pyocyanin is extracted from one of them. Cells are collected from the other half and used to quantify total protein yield and normalize the estimated corresponding pyocyanin quantities.
Keywords: PyocyaninBackground
Pyocyanin is a blue redox-active phenazine pigment produced by a human pathogen, P. aeruginosa. Pyocyanin is present in large quantities in the sputum of cystic fibrosis patients infected by P. aeruginosa (Wilson et al., 1988) and plays a major role in the virulence of the pathogen (Lau et al., 2004). Pyocyanin production is regulated by quorum sensing, which involves a cell-density-dependent synthesis of signal molecules that modulate the expression of virulence genes (Fuqua et al., 2001). Swarming is a complex communal behavior developing in response to multiple environmental signals and enabling cell motility on a semi-solid surface and colonization of host tissues. Our earlier observations suggested that pyocyanin production in liquid cultures of P. aeruginosa differ significantly from that in swarming colonies, likely due to quorum sensing regulation. Therefore, we developed a protocol allowing extraction and quantification of pyocyanin secreted by swarming cells into the surrounding agar. The obtained quantities are normalized by total cellular protein extracted from the swarming cells. This protocol differs from commonly used quantification of pyocyanin extracted from liquid cultures (Frank and Demoss, 1959) cells scrapped from a solid agar surface (De Vleesschauwer et al., 2006), or agar itself, but without normalization per total cell protein (Gallagher and Manoil, 2001). Normalized quantification of pyocyanin from swarming colonies allows studying the regulation of pyocyanin production and secretion during swarming, which is an essential component of P. aeruginosa adaptation to a host environment.
Materials and Reagents
- Glass test tubes (VWR, catalog number: 47729-578 )
- Razor blade
- Plastic storage containers and spatulas
- Glass scintillation vials (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: B7999-6 )
- Glass Pasteur pipette
- 96-well flat bottom plates (Greiner Bio One, catalog number: 655-101 )
- 50 ml Falcon tubes (VWR, catalog number: 89039-658 )
- Whatman filter paper No.1 (GE Healthcare, catalog number: 1001-090 )
- 50 ml centrifuge vials
- 1.5 ml microcentrifuge tubes
- 0.2 μm filter
- 10 ml syringes
- PFTE lined caps for glass vials (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: B7815-24 )
- Pseudomonas aeruginosa PAO1
- Chloroform (Pharmco-AAPER, catalog number: 3090000DIS )
- HCl (Pharmco-AAPER, catalog number: 284000ACS )
- Sodium hydroxide (NaOH) (Thermo Fisher Scientific, Fischer Scientific, catalog number: 1310-73-2 )
- Bradford reagent (Alfa Aesar, catalog number: J61522-AP )
- Bovine serum albumin (BSA) (Akron Biotech, catalog number: AK8917-1000 )
- Monosodium glutamate (Sigma-Aldrich, catalog number: 1446600 )
- Glycerol (Thermo Fisher Scientific, Fisher Scientific, catalog number: G33 )
- Sodium phosphate monobasic dihydrate (NaH2PO4) (Sigma-Aldrich, catalog number: 71500 )
- Potassium phosphate dibasic (K2HPO4) (Sigma-Aldrich, catalog number: 17835 )
- Sodium chloride (NaCl) (Sigma-Aldrich, catalog number: S7653 or VWR, catalog number: E529-500ML )
- Biotin (Gold Bio, catalog number: B-950-1 )
- Thiamine HCl (Gold Bio, catalog number: T-260-25 )
- Copper(II) sulfate pentahydrate (CuSO4·5H2O) (Sigma-Aldrich, catalog number: 209198 )
- Zinc sulfate heptahydrate (ZnSO4·7H2O) (Sigma-Aldrich, catalog number: Z0251 )
- Iron(II) sulfate heptahydrate (FeSO4·7H2O) (Sigma-Aldrich, catalog number: 215422 )
- Manganese(II) chloride tetrahydrate (MnCl2·4H2O) (Avantor Performance Materials, J.T. Baker, catalog number: 2540-04 )
- Yeast extract (BD, Bacto, catalog number: 212750 )
- Tryptone (BD, Bacto, catalog number: 211705 )
- Agar (BD, Bacto, catalog number: 214010 )
- Glucose (Alfa Aesar, catalog number: A16828 )
- Casamino acids (Teknova, catalog number: C2000 )
- Magnesium sulfate heptahydrate (MgSO4·7H2O) (Sigma-Aldrich, catalog number: 230391 )
- Biofilm mineral medium (BMM) (see Recipes)
- 10x basal salt solution
- Vitamin solution
- Trace metals solution
- Luria Bertani (LB) agar plates (see Recipes)
- Swarming agar plates (see Recipes)
- Potassium phosphate buffer
- 1 mM FeSO4
- 20 mM MgSO4
Equipment
- Potato masher (see Notes)
- BioMateTM 3 spectrophotometer (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 335904P )
Note: This product has been discontinued. A similar model is BioMateTM 3S spectrophotometer (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 840208400 ). - MaxQ 4000 table top shake incubator (Thermo Fisher Scientific, Thermo ScientificTM, model: SHKA4000 )
- SynergyTM Mx 2 Multi-Mode plate reader (BioTek Instruments) with Gen5TM 2.05 PC software (BioTek Instruments)
- Fume hood
- Centrifuge (Thermo Fisher Scientific, Thermo ScientificTM, model: SorvallTM ST 40R ) with 50 ml bucket insert (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 75003674 )
- Water bath sonicator (Thomas Scientific, Branson®, catalog number: 1207K40 )
- Centrifuge (Thermo Fisher Scientific, Thermo ScientificTM, model: SorvallTM RC 6 Plus Centrifuge ) with rotor type (Thermo Fisher Scientific, Thermo ScientificTM, model: F13-14x50 cy )
- Centrifuge (Eppendorf, model: 5424 )
- Table top standard orbital shaker (VWR, model: 3500 )
- 1 L graduated cylinder
Procedure
- Pyocyanin extraction
- Inoculate Pseudomonas aeruginosa strain PAO1 from frozen stock on an LB agar plate and incubate for 24 h at 37 °C.
- Pipette 5 ml of BMM into 3 sterile test tubes, inoculate isolated colonies of PAO1 grown on LB agar, and incubate for 16 h at 37 °C with shaking at 200 rpm.
- Measure OD600 (BioMate3 spectrophotometer) of the 3 biological replicates and normalize cell cultures to OD600 = 0.3 by diluting with fresh BMM.
- Inoculate 2 μl of normalized culture onto the center of a freshly made (3-4 h after pouring) swarming plate (described in Recipes section) and incubate for 24 h at 37 °C.
Cut each swarming colony together with the agar it occupies in half using a razor blade. Transfer ½ of the swarming colony into a small plastic container using a plastic spatula (Figure 1) and add 5 ml of saline (0.85% NaCl).
Figure 1. Transferring ½ of the swarming colony with the agar it occupies into a plastic container - Using a potato masher, mash the agar into small pieces (Figure 2) and transfer the agar along with saline into a 40 ml glass scintillation vial (glass separation funnel can be used instead). Repeat for two other replicates.
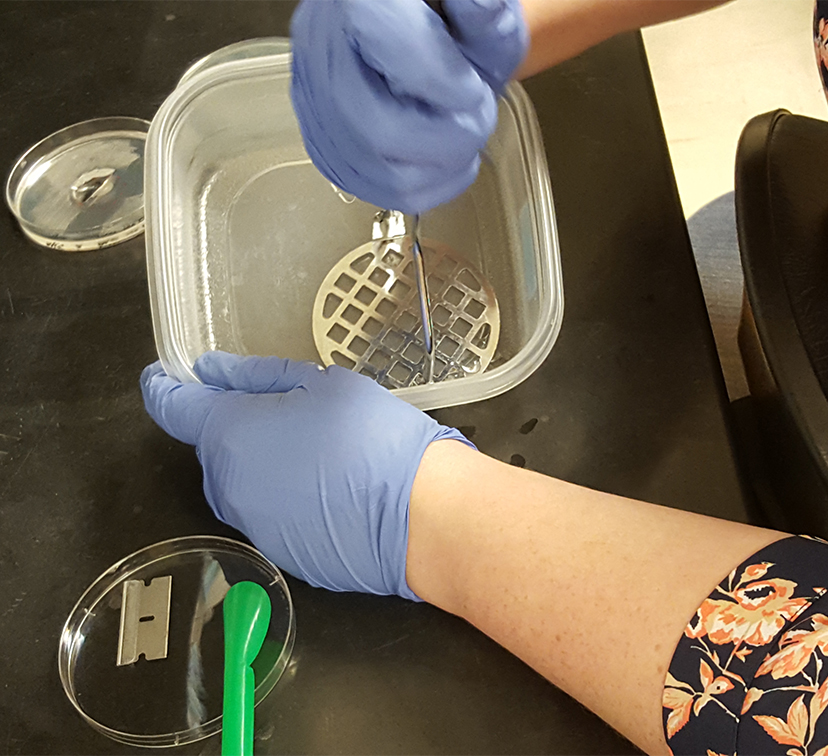
Figure 2. Smashing the collected swarming colony and agar into small pieces in preparation for extraction - Rinse the container and the masher with another 5 ml of saline, collecting all the remaining pieces of agar, and combine with the first portion containing the smashed agar.
- Using a glass pipette, add 5 ml of chloroform to the glass vials containing smashed agar. Secure the glass vials to the table top orbital shaker and shake the vials at speed setting 3 for 15 min at room temperature. To avoid the toxic effect of chloroform, all the steps involving the solvent should be done in a fume hood.
- After shaking, stand the vials vertically to let the layers separate. This will take about 10 min.
- By using a glass Pasteur pipette, transfer the bottom chloroform layer into a clean scintillation vial.
Note: If pyocyanin is present, this layer will be tinted blue. - Repeat steps A7-A10 adding chloroform two more times to the glass vials containing smashed agar and combine all three chloroform fractions for each replicate in one vial.
- Add 5 ml of 0.2 N HCl and shake the vials using a table top orbital shaker at speed setting 3 for 15 min at room temperature.
- After shaking, stand the vials vertically to let the layers separate. This will take about 10 min.
- By using a glass Pasteur pipette, collect the top HCl layer and transfer 200 μl of it to a flat bottom 96-well plate.
Note: If pyocyanin is present, this layer will be tinted red. Measure absorbance at 520 nm (SynergyTM Mx Biotek plate reader). - Calculate the concentration of pyocyanin in μg/ml by using the extinction coefficient of 17.1 (Kurachi, 1958).
- Inoculate Pseudomonas aeruginosa strain PAO1 from frozen stock on an LB agar plate and incubate for 24 h at 37 °C.
- Total cellular protein quantification
Use the remaining half of the swarming colony from step A5 of pyocyanin extraction protocol above.- Transfer the remaining ½ of the swarming colony into a small plastic container and pipette 5 ml of saline into it
- Using a potato masher, mash the agar into small pieces and transfer the agar along with saline into a 50 ml Falcon tube (Figure 2).
- Rinse the container and masher with 5 ml of saline to collect all the remaining pieces of agar and transfer to the Falcon tube used in the previous step, thus combining all the pieces of the mashed agar.
- Sonicate the mixture for 15 min in a Branson 2800 water-bath sonicator (with default setting), then vortex for 5 min. Repeat this step two more times.
- Gently pellet agar pieces by centrifuging for 2 min at 1,000 x g, carefully collect the supernatant without disturbing the agar, and transfer into a clean 50 ml Falcon tube.
- Add 10 ml of saline to agar pellet and repeat steps B4-B5. Add another 10 ml and repeat steps B4-B5. Combine all the collected supernatants and filter through a Whatman No. 1 filter to remove any leftover agar pieces. Apply vacuum if needed. Transfer the filtered supernatant into a clean 50 ml centrifuge vial.
- Centrifuge the filtrate at 17,000 x g for 10 min at 25 °C (Sorvall RC 6+ centrifuge with rotor type F13-14x50 cy). Collect the pellet, resuspend it in 1 ml of 1 N NaOH, and transfer into a 1.5 ml microfuge tube.
- Heat the samples at 90 °C for 10 min and centrifuge for 5 min at 15,000 x g (Eppendorf 5424 centrifuge) at room temperature.
- Transfer the supernatants to a new microfuge tube for Bradford analysis. Depending on the protein yield, samples may need to be diluted. In our experiments, we diluted samples 10 times with saline.
- Take 20 μl of each sample and mix with 200 μl of Bradford reagent in a 96-well flat bottom plate. Assay Bovine Serum Albumin protein standards in the same plate. Measure absorbance at 595 nm (SynergyTM Mx Biotech plate reader). Calculate protein concentration according to the BSA standard curve and use it to normalize the pyocyanin data.
- Transfer the remaining ½ of the swarming colony into a small plastic container and pipette 5 ml of saline into it
Data analysis
Total amount of pyocyanin secreted by one half of the colony was determined (Figure 3B) and normalized by the total amount of cellular protein extracted from another half of the colony (Figures 3C and 3D). To calculate total protein, we used a calibration curve based on BSA standards plotted in Microsoft Excel. Only standard curves that yielded an R2 value above 0.95 were used. The measured and normalized production of pyocyanin correlates well with the visual observation of the mutant carP::Tn5 showing significantly reduced pyocyanin production in comparison to PAO1 swarming colonies. To test the accuracy and consistency of the measurements, the experiment was repeated three times, with three biological replicates each time. Standard deviation between biological replicates was below 11 %. We observed no additional deviation due to differences in the sizes of swarming colonies.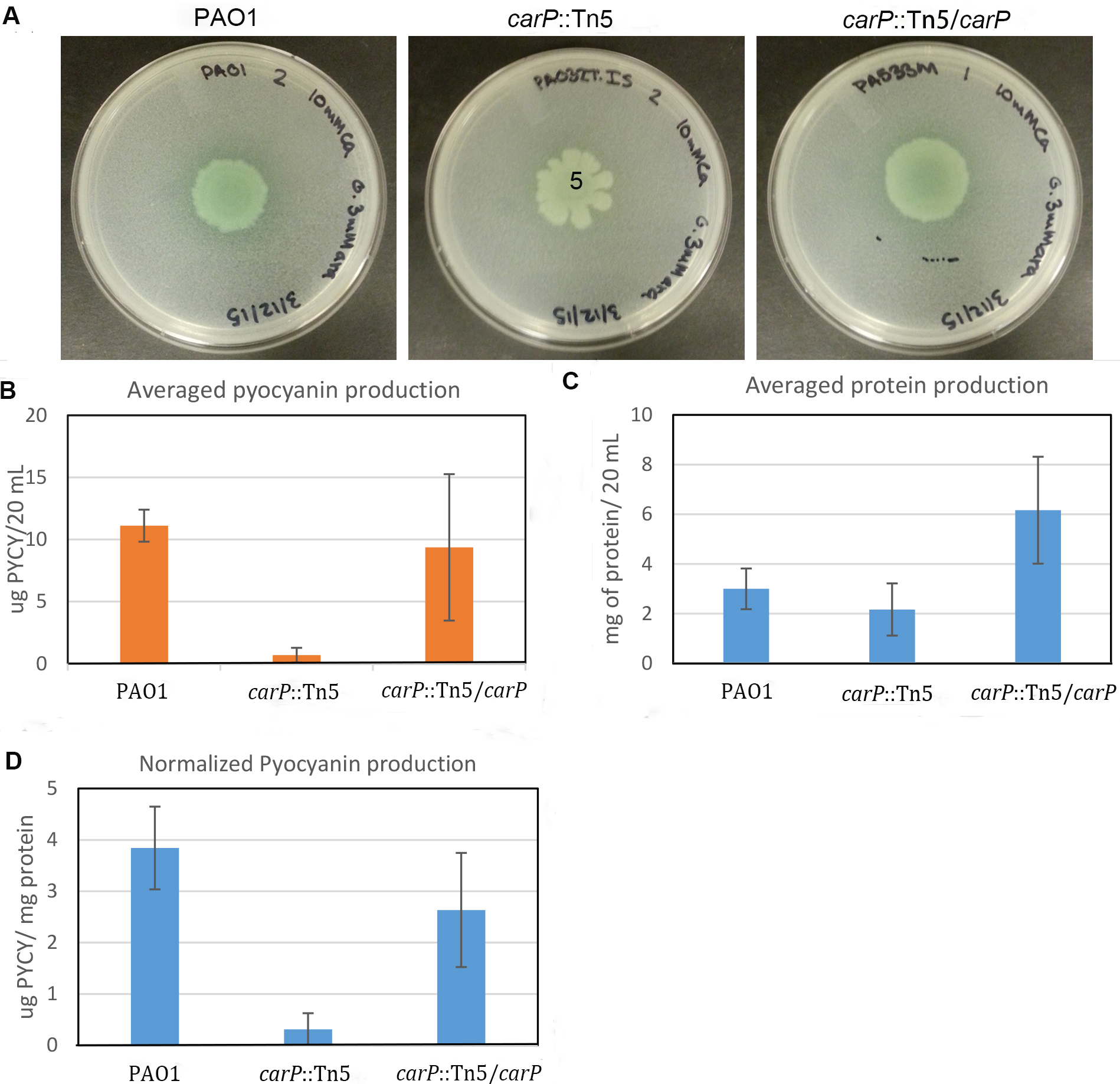
Figure 3. Quantification of pyocyanin production in P. aeruginosa swarming colonies. A. Photographs of swarming colonies formed by P. aeruginosa PAO1 and mutants with transposon disrupted (carP::Tn5) and in-trans complemented (carP::Tn5/carP) carP, encoding calcium-regulated beta-propeller protein (Guragain et al., 2016). Swarming medium [Recipes below] was complemented with 10 mM CaCl2. B. Amount of pyocyanin measured. C. Amount of protein quantified using the Bradford assay. D. Quantified pyocyanin normalized by total cellular protein.
Notes
- To enable accurate normalization of pyocyanin data, swarming colonies were cut in half, so one half can be used to measure pyocyanin, and the other to measure cell protein. We also tested an alternative approach: inoculating 2 separate plates and using one entire colony (or plate) for pyocyanin and the other colony (or plate) for protein quantification. However, colonies vary in diameters, and this prevented accurate normalization.
- The circular potato masher with 8 x 8 mm evenly spaced openings was used for convenient breaking agar into approximately even pieces.
- A non-inoculated swarming agar plate was used as a control to verify that the agar components do not interfere with pyocyanin and protein extraction and quantification.
- The number and the duration of sonication, vortexing, and extraction steps were optimized to allow maximum possible removal of swarming cells embedded into the agar, and efficient extraction of pyocyanin.
- 50 ml Falcon tubes were used for collecting cells for protein quantification, because the clear walls made it easy to visualize agar pellet. The latter was centrifuged at a low speed for a brief period to pellet only agar, but not cells. To pellet cells for the following lysis, samples were transferred into 50 ml centrifuge vials and centrifuged at 17,000 x g.
- Filtering the collected supernatant from step B6 removed all the agar pieces that were accidentally collected. If agar is not removed completely, it can interfere with cell lysis by NaOH.
Recipes
- Biofilm minimal media (BMM) (Sarkisova et al., 2005).
Mix 100 ml of sterile 10x basal salt solution (Recipe 1a) to 900 ml of sterile nanopure water. Add 1 ml of vitamin solution (Recipe 1b), 200 μl of trace metals solution (Recipe 1c), and 20 μl of 1 M MgSO4. - 10x basal salt solution
In 1 L of nanopure water combine:
15 g monosodium glutamate
46 g glycerol
0.18 g sodium phosphate monobasic dihydrate (NaH2PO4)
0.78 g potassium phosphate dibasic (K2HPO4)
84.7 g sodium chloride (NaCl)
Adjust the pH to 7.0. Sterilize by autoclaving - Vitamin solution
Dissolve 1 mg of biotin in 10 ml of nanopure water. Aliquot 1 ml of biotin stock solution in fresh tube, add 50 mg thiamine HCl to it, and mix properly. Adjust the final volume to 100 ml with nanopure water. Filter sterilize using a 0.2 μm filter and store at 4 °C. - Trace metals solution
Dilute 10 ml concentrated HCl in 70 ml of nanopure water. Add the following ingredients:
0.5 g CuSO4·5H2O
0.5 g ZnSO4·7H2O
0.5 g FeSO4·7H2O
0.2 g MnCl2·4H2O
Dissolve completely and adjust the final volume to 100 ml with nanopure water. Filter-sterilize using a 0.2 μm filter. - Luria Bertani (LB) agar plates
Combine the below ingredients in 1 L of nanopure H2O and autoclave
5 g yeast extract
5 g NaCl
10 g tryptone
15 g BD BactoTM agar - Swarming agar plates (modified from [Overhage et al., 2007])
For 1 L of media add (freshly made):
3.6 g glucose
5 g casamino acids
100 ml potassium phosphate buffer (Recipe 3a)
5 g BD BactoTM agar
Autoclave for 20 min at 250 F and 20 PSI
After autoclaving add:
1 ml of 20 mM MgSO4 (final concentration = 0.02 mM) (Recipe 3b)
10 ml of 1 mM FeSO4 (final concentration = 0.01 mM) (Recipe 3c)
Using a 25 ml glass pipette, pipette 20 ml of medium into every plate. Then place the plates individually on a bench top to dry at room temperature for 3-4 h before inoculation. - Potassium phosphate buffer
Make 1 L of K2HPO4 (F.W:174.18). Add 107.99 g/1 L of nanopure H2O
Make 1 L of KH2PO4 (F.W: 136.09). Add 84.37 g/1 L of nanopure H2O
In a 1 L graduated cylinder, mix:
615 ml 620 mM K2HPO4
385 ml 620 mM KH2PO4
Buffer pH should be 7 - 1 mM FeSO4
To make 1 mM stock solutions add 0.0559 g of FeSO4 to 200 ml nanopure H2O. Filter sterilize and store at 4 °C - 20 mM MgSO4
To make 20 mM stock solutions add 0.9858 g of MgSO4 to 200 ml nanopure H2O. Filter sterilize and store at 4 °C
Acknowledgments
We thank Dr. Erica Lutter, Oklahoma State University, for sharing equipment and for helpful discussions. This work was supported by the Research Grant from OCAST (Award HR12-167).
References
- De Vleesschauwer, D., Cornelis, P. and Hofte, M. (2006). Redox-active pyocyanin secreted by Pseudomonas aeruginosa 7NSK2 triggers systemic resistance to Magnaporthe grisea but enhances Rhizoctonia solani susceptibility in rice. Mol Plant Microbe Interact 19(12): 1406-1419.
- Frank, L. H. and Demoss, R. D. (1959). On the biosynthesis of pyocyanine. J Bacteriol 77(6): 776-782.
- Fuqua, C., Parsek, M. R. and Greenberg, E. P. (2001). Regulation of gene expression by cell-to-cell communication: acyl-homoserine lactone quorum sensing. Annu Rev Genet 35: 439-468.
- Gallagher, L. A. and Manoil, C. (2001). Pseudomonas aeruginosa PAO1 kills Caenorhabditis elegans by cyanide poisoning. J Bacteriol 183(21): 6207-6214.
- Guragain, M., King, M. M., Williamson, K. S., Perez-Osorio, A. C., Akiyama, T., Khanam, S., Patrauchan, M. A. and Franklin, M. J. (2016). The Pseudomonas aeruginosa PAO1 two-component regulator CarSR regulates Calcium homeostasis and Calcium-induced virulence factor production through its regulatory targets CarO and CarP. J Bacteriol 198(6): 951-963.
- Kurachi, M. (1958). Studies on the biosynthesis of pyocyanine. (ii) : isolation and determination of pyocyanine. Bulletin of the Institute for Chemical Research, Kyoto University 36(6): 776-782.
- Lau, G. W., Hassett, D. J., Ran, H. and Kong, F. (2004). The role of pyocyanin in Pseudomonas aeruginosa infection. Trends Mol Med 10(12): 599-606.
- Overhage, J., Lewenza, S., Marr, A. K. and Hancock, R. E. (2007). Identification of genes involved in swarming motility using a Pseudomonas aeruginosa PAO1 mini-Tn5-lux mutant library. J Bacteriol 189(5): 2164-2169.
- Sarkisova, S., Patrauchan, M. A., Berglund, D., Nivens, D. E. and Franklin, M. J. (2005). Calcium-induced virulence factors associated with the extracellular matrix of mucoid Pseudomonas aeruginosa biofilms. J Bacteriol 187(13): 4327-4337.
- Wilson, R., Sykes, D. A., Watson, D., Rutman, A., Taylor, G. W. and Cole, P. J. (1988). Measurement of Pseudomonas aeruginosa phenazine pigments in sputum and assessment of their contribution to sputum sol toxicity for respiratory epithelium. Infect Immun 56(9): 2515-2517.
Article Information
Copyright
© 2016 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
King, M. M., Guragain, M., Sarkisova, S. A. and Patrauchan, M. A. (2016). Pyocyanin Extraction and Quantitative Analysis in Swarming Pseudomonas aeruginosa. Bio-protocol 6(23): e2042. DOI: 10.21769/BioProtoc.2042.
Category
Microbiology > Microbial metabolism > Other compound
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.