- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Identification of Methylated Deoxyadenosines in Genomic DNA by dA6m DNA Immunoprecipitation



Published: Vol 6, Iss 21, Nov 5, 2016 DOI: 10.21769/BioProtoc.1990 Views: 10109
Reviewed by: Gal HaimovichZhen ShiBenoit Chassaing
Original research article
The authors used this protocol in:

Jan 2016
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
dA6m DNA immunoprecipitation followed by deep sequencing (DIP-Seq) is a key tool in identifying and studying the genome-wide distribution of N6-methyldeoxyadenosine (dA6m). The precise function of this novel DNA modification remains to be fully elucidated, but it is known to be absent from transcriptional start sites and excluded from exons, suggesting a role in transcriptional regulation (Koziol et al., 2015). Importantly, its existence suggests that DNA might be more diverse than previously believed, as further DNA modifications might exist in eukaryotic DNA (Koziol et al., 2015). This protocol describes the method to perform dA6m DNA immunoprecipitation (DIP), as was applied to characterize the first dA6m methylome analysis in higher eukaryotes (Koziol et al., 2015). In this protocol, we describe how genomic DNA is isolated, fragmented and then DNA containing dA6m is pulled down with an antibody that recognizes dA6m in genomic DNA. After subsequent washes, DNA fragments that do not contain dA6m are eliminated, and the dA6m containing fragments are eluted from the antibody in order to be processed further for subsequent analyses.
Background
This protocol was developed in order to identify regions in the genome that contain dA6m. It can be used to detect dA6m in different genomes. As a guideline, this protocol was established from existing approaches used to detect adenosine methylation in RNA (Dominissini et al., 2013). We developed this protocol and adapted it for the detection of dA6m in DNA, rather than detecting adenosine methylation RNA. This was required, as no protocol was available at that time to allow the genome-wide identification of dA6m in eukaryotic DNA.
Materials and Reagents
- Microcentrifuge tubes, 1.7 ml (Coring, Axygen®, catalog number: MCT-175-C )
- 1.5 ml Bioruptor Pico microtubes with caps (Diagenode, catalog number: C30010016 )
- Optional: D1000 tape and reagents (Agilent Technologies, catalog numbers: 5067-5582 ; 5067-5583 )
- DNeasy Blood & Tissue Kit (QIAGEN, catalog number: 69506 )
- Phosphate-buffered saline (PBS)
- CutSmart® buffer (New England Biolabs, catalog number: B7204S )
- RNase (DNase-free) (Roche Diagnostics, catalog number: 11119915001 )
- Water (double deionized water that has been autoclaved)
Note: All water used in this protocol refers to double deionized water, which has been subsequently autoclaved. As an alternative, purchased DNAse free water could be used. - Qubit® dsDNA HS Assay Kit (Thermo Fisher Scientific, InvitrogenTM, catalog number: Q32854 )
- Bovine serum albumin (BSA) (Sigma-Aldrich, catalog number: A3059 )
- dA6m antibodies (Synaptic Systems, catalog numbers: 202011 / 202111 / 202003 )
- Dynabeads® protein A (Thermo Fisher Scientific, NovexTM, catalog number: 10008D )
- Glycogen (Roche Diagnostics, catalog number: 10901393001 )
- NaOAc (Thermo Fisher Scientific, Fisher Scientific, catalog number: 10010500 )
- Isopropanol (Thermo Fisher Scientific, Fisher Scientific, catalog number: 10315720 )
- Ethanol absolute (VWR, catalog number: 20821330 )
- TruSeq Nano DNA LT Library Preparation Kit (set A or B) (Illumina, catalog number: FC-121-4001 or FC-121-4002 )
- Tris-HCl (Melford Laboratories, catalog number: B2005 )
- Sodium chloride (NaCl) (Thermo Fisher Scientific, Fisher Scientific, catalog number: 10428420 )
- Igepal CA-630 (Sigma-Aldrich, catalog number: I8896 )
- N6-methyladenosine 5’-monophosphate sodium salt (Sigma-Aldrich, catalog number: M2780 )
- EDTA (Thermo Fisher Scientific, Fisher Scientific, catalog number: 10213570 )
- Dry ice
- 5x DIP IP buffer (see Recipes)
- 1x DIP IP buffer (see Recipes)
- DIP elution buffer (see Recipes)
Equipment
- Incubator (Grant, dry block heating system)
- Rotating wheel (Bibby Scientific, Stuart, model: rotator SB3 )
- Thermomixer (Eppendorf, model: Thermomixer compact )
- Qubit fluorometer (Thermo Fisher Scientific, InvitrogenTM, catalog number: Q32857 )
Note: This product has been discontinued. - Water bath sonicator (Diagenode, Bioruptor Pico, catalog number: B01060001 )
- Microcentrifuge (Eppendorf, model: Centrifuge 5424 )
- Vortex (Scientific Industries, model: Vortex-Genie 2 )
- Magnet (Thermo Fisher Scientific, DynaMagTM, catalog number: 12321D )
- Optional: 2200 Tapestation (Agilent Technologies, catalog number: G2965A )
Procedure
The procedure can be divided into three parts: ‘A’ isolation of genomic DNA, ‘B’ sonication and ‘C’ DNA immunoprecipitation of dA6m. An outline of the procedure is illustrated in Figure 1. We encourage carrying out all experiments with at least two biological replicates, and with at least 2 different dA6m antibodies. As a further control, one could carry out the DNA immunoprecipitation procedure on E. coli bacterial DNA, as described in the original work (Koziol et al., 2015). Wild type bacteria could be used as a positive control, as their dA6m sites are well known. As a negative control, bacteria deficient in the Dam dA6m methylase can be used. Such bacteria no longer have deoxyadenosine methylation within the sequence GATC, and overall have a reduced number of dA6m sites. The success of the DNA immunoprecipitation of dA6m can then be accessed by observing a significant difference between both bacterial genomes in their dA6m signal, specifically by the reduction in dA6m within GATC sequences (Koziol et al, 2015).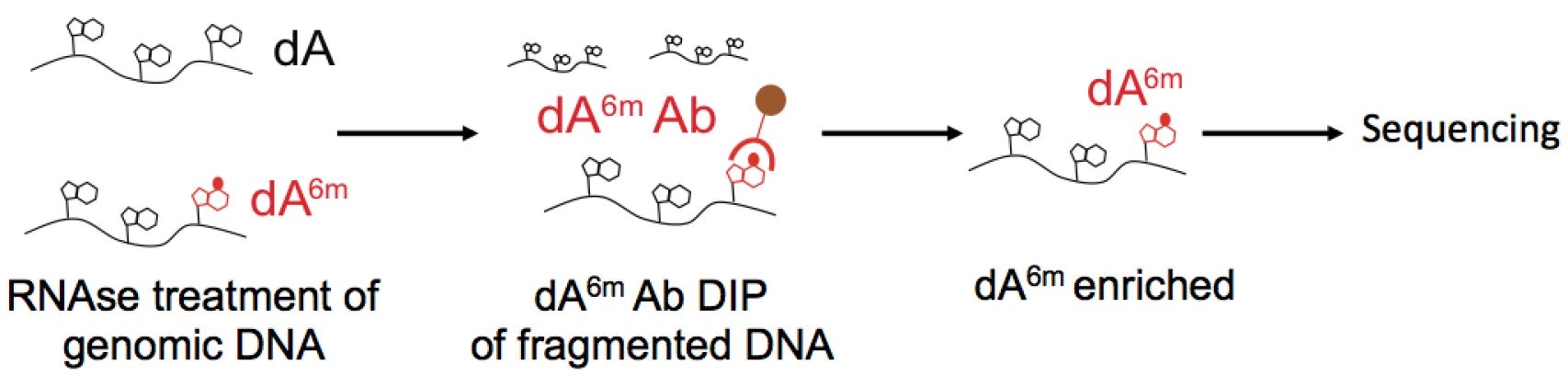
Figure 1. Illustration of dA6m DNA immunoprecipitation. Genomic DNA is isolated and any RNAs are eliminated by RNase treatment. Next, the DNA is fragmented and enriched by DNA immunoprecipitation using an antibody against dA6m. The enriched fraction can then be analyzed by subsequent deep sequencing.
- Genomic DNA isolation
- Extract genomic DNA using the DNeasy Blood & Tissue Kit, following the manufacturer’s guidelines, but with the exception: elute the DNA with 177 μl of PBS, instead of AE buffer (10 mM Tris-Cl, 0.5 mM EDTA, pH 9.0). Next, add again 177 μl of PBS onto the column, and elute. Collect both fractions in the same tube, making a total of 354 μl.
Note: Any method to extract genomic DNA can be used in this step, as long as at least 5 μg of DNA is obtained. If another method of genomic DNA extraction is used, ensure you dilute your genomic DNA so that the final volume is 354 μl. In this volume, the concentration of DNA should be at least 14 ng/μl. - Add 40 μl of CutSmart buffer.
- Add 6 μl of RNase DNase-free.
- Incubate at 37 °C overnight.
Note: Incubate for at least 6 h to ensure a good degradation of RNA. RNAs contain methylated adenosines, which the antibody can also recognize. Hence, the presence of RNA might inhibit recovery of methylated deoxyadenosines from DNA making removal important. - Split the sample into two, and clean up the genomic DNA: Use the DNeasy Blood & Tissue Kit, following the manufacturer’s protocol ‘DNeasy Blood & Tissue Kit’, eluting the DNA once with 177 μl of PBS, instead of AE buffer.
Note: This column cleanup step helps to better eliminate the RNAs from the sample. The split into two columns is required as otherwise the volumes that required to be loaded exceed the column capacity. - Combine the two corresponding samples.
- Add 40 μl of CutSmart buffer.
- Add 2 μl of RNase DNase-free.
- Incubate at 37 °C for at least 2 h.
Note: This additional RNase step ensures that most of the RNA is degraded. - Divide the sample in two, and clean up the genomic DNA: Use the DNeasy Blood & Tissue Kit, following the manufacturer’s guidelines, eluting the DNA once with 200 μl of water, instead of AE buffer.
Note: The split into two columns is required as otherwise the volumes that required to be loaded exceed the column capacity. - Combine the two corresponding samples.
- Quantify genomic DNA content using the Qubit Fluorometer and the Qubit dsDNA HS Assay Kit, by following the manufacturer’s guidelines.
Note: We recommend not to use Nanodrop for quantification, as this does not result in reliable quantification. At this point, the protocol can be stopped and the DNA can be frozen. We recommend storage at -20 °C or colder.
- Extract genomic DNA using the DNeasy Blood & Tissue Kit, following the manufacturer’s guidelines, but with the exception: elute the DNA with 177 μl of PBS, instead of AE buffer (10 mM Tris-Cl, 0.5 mM EDTA, pH 9.0). Next, add again 177 μl of PBS onto the column, and elute. Collect both fractions in the same tube, making a total of 354 μl.
- Sonication of genomic DNA
- Take 5 μg of genomic DNA and transfer into a 1.5 ml Bioruptor Pico microtube.
- Adjust the volume to 300 μl with water.
- Sonicate in Diagenode Bioruptor Pico for 35 cycles, 30 sec on/30 sec off, at 4 °C. A representative example is shown in Figure 2.
Note: Sonication time needs to be optimized depending on the machine and genomic DNA used. We aim for a DNA fragment size that peaks at about 150-300 base pairs. - Verify fragmentation of genomic DNA with the Tapestation, using the D1000 tape and reagents, following the manufacturer’s protocol.
Note: Alternatively to the Tapestation, genomic DNA can be run on an agarose gel to verify the fragment size following sonication.
- Take 5 μg of genomic DNA and transfer into a 1.5 ml Bioruptor Pico microtube.
- dA6m DNA immunoprecipitation
- Take aside 300 ng of sonicated genomic DNA for each sample. This will be the input control for deep sequencing and will not be used for the DNA immunoprecipitation procedure described below. Store at -20 °C until needed.
- Take 3 μg of the sonicated genomic DNA, and add to a 1.7 ml microcentrifuge tube.
- Place the tube on ice.
- On ice, add 200 μl of 5x DIP IP buffer.
- On ice, add 200 μl of 30 μg/μl BSA stock solution.
- On ice, adjust the total volume to 1 ml with water.
- Vortex briefly, about 2 sec, and place on ice.
- Add 5 μl of 0.5 μg/μl dA6m antibody stock.
- Immediately mix gently by turning the tube and place the tube back on ice.
- Mix magnetic protein A beads by vortexing, so that all beads are resuspended.
- Take 100 μl of mixed magnetic protein A beads per sample, and add to a new 1.7 ml microcentrifuge tube.
- Place on magnet and wait for 1 min for magnetic beads to stick to the side of tube.
- Remove the supernatant from the beads.
- Remove beads from magnet.
- Add 1 ml of 1x DIP IP buffer to the beads and mix gently.
- Repeat steps C11-C14 at least two more times.
- Place on magnet and wait for 1 min for magnetic beads to stick to the side of tube.
- Remove the supernatant from the beads.
- Resuspend beads in 105 μl 1x DIP IP buffer.
- Add 105 μl of 30 μg/μl BSA stock solution.
- Incubate the beads separately on a rotating wheel at 4 °C.
Note: The beads and samples are incubated in separate tubes. - Incubate the samples separately on a rotating wheel at 4 °C.
- After an overnight incubation, add 200 μl of the bead suspension to each sample.
- Incubate the sample-bead mixture for at least one more hour on the rotating wheel at 4 °C.
- Place sample on magnet and wait for 1min for magnetic beads to stick to the side of tube.
- Remove all the supernatant from the beads.
- Remove beads from magnet.
- Add 1 ml of 1x DIP IP buffer to the beads and mix gently.
- Repeat steps C23-C26 at least three more times.
- Place sample on magnet and wait for 1 min for magnetic beads to stick to the side of tube.
- Remove all supernatant from the beads.
- Add 200 μl of the DIP elution buffer.
- Vortex.
- Place sample in Thermomixer at 42 °C for at least 1 h, at 1,400 rpm.
- Spin the samples down.
- Place sample on magnet and wait for 1min for magnetic beads to stick to the side of tube.
- Transfer the supernatant from the beads into a new 1.7 ml microcentrifuge tube.
Note: You can discard the beads. - Add 300 μl of water to the recovered supernatant.
- Add 2 μl glycogen.
- Add 50 μl of 3 M stock of NaOAc.
- Add 500 μl isopropanol.
- Vortex.
- Place on dry ice until frozen.
- Centrifuge at maximum speed for 30 min.
Note: A pellet should have formed, that contains the dA6m enriched DNA. - Remove supernatant.
- Wash pellet twice with 1 ml of 70% ethanol.
- Remove any liquid.
- Let pellet dry for 5 min at room temperature.
- Resuspend in desired amount of water.
Note: We usually resuspend in about 10-50 μl. - Quantify genomic DNA content using the Qubit Fluorometer and the Qubit dsDNA HS Assay Kit, by following the manufacturer’s guidelines.
Note: This step is required to ensure that you have actually obtained genomic DNA with the pulldown. We usually expect to obtain at least 10 ng of genomic DNA. - Use all genomic DNA from step C47 and the corresponding input control from step C1 to make standard deep sequencing libraries.
Note: Although any standard deep sequencing is possible, we recommend using the TruSeq Nano DNA LT Library Preparation Kit (set A or B) for preparation of libraries suitable for Illumina sequencing with for example HiSeq 2500 or HiSeq 4000.
- Take aside 300 ng of sonicated genomic DNA for each sample. This will be the input control for deep sequencing and will not be used for the DNA immunoprecipitation procedure described below. Store at -20 °C until needed.
Data analysis
Any information about data processing and analysis, replicates, independent experiments, reproducibility, including any statistical tests applied, has been provided in the original research paper where this method has been applied (Koziol et al., 2015).
Representative data
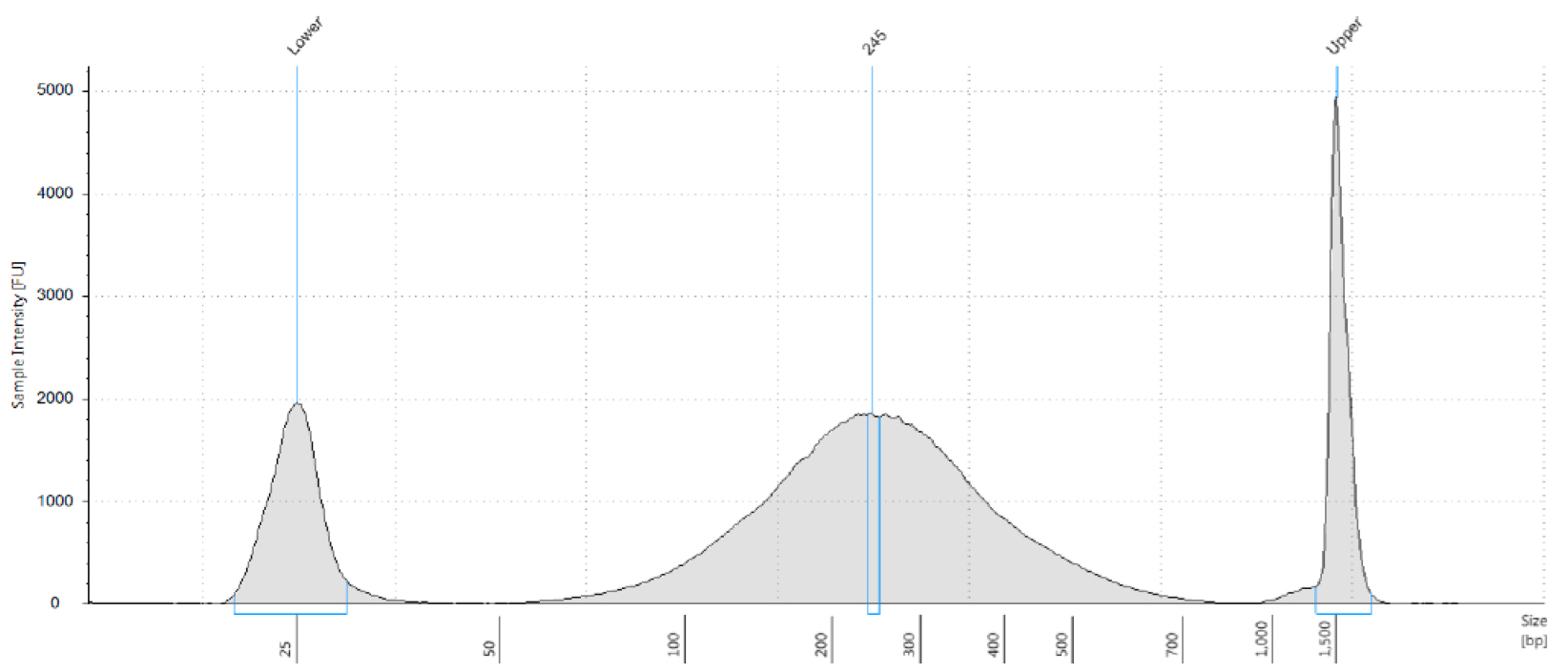
Figure 2. Distribution of genomic DNA fragmentation pattern after sonication. 5 μg of genomic DNA was fragmented in a volume of 300 μl for 35 min with the Diagenode sonicator. The size of the genomic DNA was determined by Tapestation analysis, with the D1000 Tapestation reagents. In this sample, the peak of the fragmented DNA was determined to be 245 bp. Lower and upper markers are also shown.
Notes
- We have found that this protocol is very robust and reproducible. We were always able to enrich for dA6m and always obtained sufficient DNA for subsequent standard deep sequencing libraries. If the method does not result in enough or no DNA, which we did not experience, repeat the experiment again, maybe with a fresh antibody batch.
- We tested 3 different dA6m antibodies for the enrichment of dA6m in the eukaryotic genome. All of which are listed above. We have found that all of them gave reproducible and overlapping results (Koziol et al., 2015). Hence, we believe any of these antibodies will work. However, we encourage confirming any initial results with at least two different dA6m antibodies, to have a higher confidence in the results.
Recipes
Note: Prepare all the solutions fresh from stock solutions before starting and keep on ice.
- 5x DIP IP buffer
Add indicated amount to 7.5 ml water
0.5 ml of 1 M Tris-HCl (pH 7.4) stock
1.5 ml of 5 M NaCl stock
0.5 ml of 10% vol/vol Igepal CA-630 stock - 1x DIP IP buffer
Mix 10 ml of 5x DIP IP buffer with 40 ml of water - DIP elution buffer
Add indicated amount for each sample, scale up according to number of samples
45 μl of 5x DIP IP buffer
75 μl of 20 mM N6-methyladenosine 5’-monophosphate sodium salt stock
105 μl of water
Acknowledgments
This protocol was used for the work previously published in Nature Structural and Molecular Biology (Koziol et al., 2015). M.J.K. was supported by the Long-Term Human Frontiers Fellowship (LT000149/2010-L), the Medical Research Council grant (G1001690), and by the Isaac Newton Trust Fellowship (RG76588). The work was sponsored by the Biotechnology and Biological Sciences Research Council grant BB/M022994/1 (M.J.K.). The Gurdon laboratory where this work was carried out is funded by the grant 101050/Z/13/Z from the Wellcome Trust, and is supported by the Gurdon Institute core grants, namely by the Wellcome Trust Core Grant (092096/Z/10/Z) and by the Cancer Research UK Grant (C6946/A14492). C.R.B. and G.E.A. are funded by the Wellcome Trust Core Grant. We thank S. Moss and K. Harnish for their advice and assistance with deep sequencing, who were supported by both Gurdon Institute core grants. We are grateful to Nigel Garrett for his critical comments. This protocol reported here was established from existing approaches used to detect adenosine methylation in RNA (Dominissini et al., 2013).
References
- Dominissini, D., Moshkovitz, M. S., Divon, S. M., Amariglio, N. and Rechavil, G. (2013). Transcriptome-wide mapping of N6-methyladenosine by m6A-seq based on immunocapturing and massively parallel sequencing. Nature Protocols 8(1): 176-189.
- Koziol, M. J., Bradshaw, C. R., Allen, G. E., Costa, A. S., Frezza, C. and Gurdon, J. B. (2016). Identification of methylated deoxyadenosines in vertebrates reveals diversity in DNA modifications. Nat Struct Mol Biol 23(1): 24-30.
Article Information
Copyright
© 2016 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Koziol, M. J., Bradshaw, C. R., Allen, G. E., Costa, A. S. H. and Frezza, C. (2016). Identification of Methylated Deoxyadenosines in Genomic DNA by dA6m DNA Immunoprecipitation. Bio-protocol 6(21): e1990. DOI: 10.21769/BioProtoc.1990.
Category
Molecular Biology > DNA > DNA extraction
Molecular Biology > DNA > DNA modification
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.