- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Neurosphere Co-culture Assay


Published: Vol 6, Iss 15, Aug 5, 2016 DOI: 10.21769/BioProtoc.1883 Views: 14737
Reviewed by: Manuel SarmientoAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Sep 2015
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
The hippocampal niche is one of two areas in the brain where stem cells reside. In this neurogenic niche, stem cells can be found in close proximity to astrocytes and in contact with microvessels consisting of pericytes and endothelial cells. To study the regulatory interplay of this complex niche network in a simplified in vitro model, we established a co-culture system. We investigate the formation of neurosphere under different co-culture conditions by using primary niche cells. Here, we describe the isolation procedure for primary niche cells culture of astrocytes, endothelial cells and pericytes/smooth muscle cells from mouse brain. These niche cells are co-cultured (by hanging inserts) with freshly isolated stem and precursor cells from the adult hippocampus to study the influence of soluble factors. This method is used to investigate factors and cell types regulating stem cell behavior in a niche-like environment.
Keywords: NeuroscienceMaterials and Reagents
- Petri dishes (diameter 5.5 cm to 9 cm)
- Falcon tubes 50 ml and 15 ml
- Eppendorf tubes 2 ml (sterilized by autoclave)
- Pasteur pipettes (1 mm diameter)
- Brush (da Vinci, model: Nova synthetics series 1570, #3 )
- 12-well and 24-well tissue culture plates (growth-enhance treated, gamma-sterilised, free of pyrogens, free of DNA/RNA, DNase/RNase)
- Filter discs (Sartorius AG, catalog number: FT-3-205-055 )
- Falcon cell strainers 40 µm and 100 µm (Thermo Fisher Scientific, catalog number: 08-771-1 and 08-771-19 )
- Hanging insert for 24-well plates, 1 µm PET (Merck Millipore Corporation, catalog number: PIRP12R48 )
- 27G ¾ needle (Braun, StericanTM, catalog number: 4657705 )
- Mice at the age of six weeks
Note: 6-8 mice for endothelial and pericyte isolation, at least 1 mouse for neurosphere co-culture assay - Mice at the age of four weeks
Note: 4 mice for astrocyte isolation are needed for one preparation. - Distilled water
- Poly-D-lysine hydrobromide (PDL) (Sigma-Aldrich, catalog number: P7280 )
- Laminin (Sigma-Aldrich, Roche, catalog number: R11243217001 )
- 70% ethanol
- HBSS (Thermo Fisher Scientific, GibcoTM, catalog number: 14175 )
- Dextran from Leuconostoc mesenteroides average molecular weight 150,000 (Sigma-Aldrich, catalog number: D4876 )
- DMEM/F-12 without glutamine (store at 4 °C) (Thermo Fisher Scientific, GibcoTM, catalog number: 21331020 )
- DMEM/F-12 with L-glutamine and 15 mM HEPES (store at 4 °C) (Thermo Fisher Scientific, GibcoTM, catalog number: 11330032 )
- B27 (Thermo Fisher Scientific, GibcoTM, catalog number: 0080085SA )
- MCDB131 Media (Thermo Fisher Scientific, catalog number: 10372-019 )
- Glutamax (100x stock) (store at 4 °C) (Thermo Fisher Scientific, CTSTM, catalog number: A12860-01 )
- Insulin-transferrin-selenium (ITS) premix (store at -20 °C) (Thermo Fischer Scientific, CorningTM, catalog number: CB-40351 )
- Non-essential amino acids (100x stock) (store at 4 °C) (Thermo Fisher Scientific, InvitrogenTM, catalog number: 11140-035 )
- Pen/Strep 100,000 U/ml (-20 °C) (Thermo Fisher Scientific, GibcoTM, catalog number: 15140 )
- Heparin (store at 4 °C) (MP Biomedicals, catalog number: 0210193125 )
- Collagenase A (Sigma-Aldrich, Roche, catalog number: 10103578001 )
- Fetal bovine serum (FBS) (-20 °C) (Merck Millipore Corporation, Biochrom, catalog number: S0613 )
- Plasma derived platelet poor serum (Sigma-Aldrich, catalog number: P2918 )
- Human EGF (store aliquots at -20 °C) (PEPROTECH, catalog number: AF100-15 )
- Human FGF-basic (store aliquots at -20 °C) (PEPROTECH, catalog number: 100-18B )
- Human VEGF (store aliquots at -20 °C) (PEPROTECH, catalog number: 100-20 )
- Accutase (store at 4 °C) (GENTAUR, PAA Laboratories GmbH, catalog number: L11-007 )
- Neuronal tissue dissociation kit (P) (Miltenyl Biotec, catalog number. 130-092-628 )
- Trypsin/EDTA (store at -20 °C) (Merck Millipore Corporation, Biochrom, catalog number: L2153 )
- PBS with 2% FBS (see Recipes)
- 30% dextran solution (see Recipes)
- Endogrow media (see Recipes)
- Astrocyte media (see Recipes)
- Pericyte/smooth muscle cell media (see Recipes)
- Neurosphere media (see Recipes)
Equipment
- Dissection tools: scissors, small spatula, fine curved forceps and scalpel (see Figure 2B)
- 7 ml Dounce tissue grinder (Capitol Scientific, Wheaton, catalog number: 357542 )
- Autoclave
- Vacuum pump
- Dounce homogenizer
- Dissection microscope (Olympus, model: SZ61 )
- Inverted microscope (Olympus, model: CKX41 )
- Scale plate insert into the eyepiece of the microscope (Olympus)
- Cooling centrifuge fitting 2 ml tubes (Eppendorf, model: 5430R )
- Swing bucket centrifuge for 15 ml tubes (Eppendorf, model: 5810R )
- Incubator at 37 °C with 5% CO2
- Tube rotator or orbital shaker
Procedure
Note: A general overview of the isolation procedure can be found in Figure 1.
- General Preparations
- At least two days prior to isolation of endothelial cells from tissue, prepare 4 poly-D-lysine (PDL)/laminin coated 12-well plates (with at least 3 wells coated per plate). To prepare wells add enough PDL (5 µg/ml in dH2O) to coat the surface and incubate overnight at room temperature. Remove the solution from the dish and wash the dish three times with dH2O. Allow to air dry. Add laminin (5 µg/ml in cold DMEM:F12) and incubate at 37 °C overnight. Remove the laminin and either use the plates immediately or store with the laminin at -20 °C until required.
- At least one day prior to astrocyte isolation prepare PDL coated plates (at least 3 wells coated per plate). Per well add enough PDL (5 µg/ml in dH2O) to coat the surface and incubate overnight at room temperature. Remove the solution from the dish and wash the dish three times with dH2O. Add dH2O and freeze at -20 °C or use directly.
- Prepare fire polished pipettes with "medium" and "small" bores by rotating glass Pasteur pipettes in a flame until the edges become rounded. We measured that our “small” bores are around 0.3-0.4 mm, whereas the “medium” pores are around 0.6-0.8 mm internal diameter. Autoclave pipettes to sterilize.
- At least two days prior to isolation of endothelial cells from tissue, prepare 4 poly-D-lysine (PDL)/laminin coated 12-well plates (with at least 3 wells coated per plate). To prepare wells add enough PDL (5 µg/ml in dH2O) to coat the surface and incubate overnight at room temperature. Remove the solution from the dish and wash the dish three times with dH2O. Allow to air dry. Add laminin (5 µg/ml in cold DMEM:F12) and incubate at 37 °C overnight. Remove the laminin and either use the plates immediately or store with the laminin at -20 °C until required.
- Isolation of microvascular endothelial cells and pericyte/vascular smooth muscle cells(vSMC)
Note: An overview of the isolation procedure can be found in Figure 1. A step-by-step picture series of the isolation procedure can be found in Figures 2 and 3.
Two weeks prior to co-culture isolate endothelial cells and pericytes/vSMC from six to eight mice at the age of 6 to 8 weeks. The protocol for endothelial cell isolation was modified from Wu et al., 2003.- Anesthetize 6-8 adult mice according to the appropriate institutional guidelines. Perform cervical dislocation.
- Spray the head with 70% ethanol to sterilize the area to minimize contamination through fur.
- With sharp scissors decapitate the mouse at the base of the brain stem.
- Cut the skin sagittal along the midline till an arbitrary point between the eyes. Expose the skull free of overlying skin and subcutaneous tissue (Figure 2C).
- Cut the skull between eyes by placing each blade of the scissors into each eye cavity. Next, make two lateral cuts at the base of the skull, following a cut through the skull along the sagittal suture.
- Expose the brain by peeling back the skull with the scissors or forceps and remove the brain from the skull with a small spatula (Figure 2D).
- Rinse the brain with PBS and transfer to a Falcon tube with PBS/2% FBS, which is kept on ice.
- After collecting all brains transfer one brain to a petri dish with a filter disc inside filled up with PBS/2% FBS.
- With the curved forceps cut of the midbrain and the cerebellum (Figure 2F).
- Remove the meninges and any visible surface blood vessels with a fine paint brush, the big vessels will get trapped easily on the filter disc (Figure 2G-H).
- With the brush remove the white matter tracts of the corpus callosum (Figure 2I).
- Transfer the tissue now to a new petri dish and keep cortex tissue on ice till further processing. Continue to process the rest of the brains in the same way (Figure 2J).
- After processing all brain samples, chop the cortex tissue into small pieces with the scalpel. Homogenize the tissue in 2 ml PBS/2% FBS using a Dounce homogenizer with a loose pestle by applying 6-8 strokes (Figures 2K-L and 3A).
- Mix the tissue homogenate (should be ~3 ml) with 3 ml of 30% dextran to get a final concentration of 15% (Figure 3B).
- Transfer the mixture into 2 ml Eppendorf tubes and centrifuge at 6,000 x g for 20 min at 4 °C (Figure 3C tube after centrifugation).
- Remove the thick supernatant carefully with a 1 ml pipette or by aspiration using a vacuum pump (Figure 3D-E). Take care to remove tissue homogenate properly from the walls. Resuspend the loose pellets in PBS with 2% FBS with a 1 ml pipette by rapidity pipetting up and down (Figure 3F).
- Place a 100 µm sterile nylon cell strainer inverse on top of a 50 ml tube. Filter the solution through the 100 µm filter and wash extensively with at least 10ml PBS containing 2% FBS. Label tube as “flow through 1” and keep on ice till further processing (Figure 3G).
- Take the 100 µm filter off and flip it into a new 50 ml tube labeled “pericytes/vSMC” (Figure 3H). Wash the filter extensively with at least 10 ml PBS containing 2% FBS (Figure 3I).
- Keep the big vessels in solution (pericytes/vSMC) on ice till centrifugation.
- Take flow through 1 and filter through a 40 µm sterile nylon cell strainer (again put it inverse on a 50 ml tube) (Figure 3J), wash with PBS containing 2% FBS to remove single cells and debris.
- The flow through can be discarded and the 40 µm nylon strainer should be flipped and inserted into a new 50 ml tube.
- Wash the nylon membrane extensively with at least 10 ml PBS containing 2% FBS to get all the microvessels back into solution, label the tube “endothelial cells”.
- Spin both tubes by centrifugation at 800 x g for 5 min at 4 °C.
- Prepare collagenase solution by dissolving 1 mg/ml collagenase A in respective growth medium endogrow and pericyte media without addition of growth factors. Dissolves best at 37 °C (see Recipes 3 and 4).
Note: Volume of collagenase solution depends on pellet size we used 3-4 ml per pellet/ cell type. - Dissolve the vessel pellet from step B23 in respective collagenase solution. Incubate the vessels in solution in a 15 ml tube for 4-5 h at 37 °C on tube rotator (Figure 3K-L). In between vigorously pipette up and down with a 1 ml pipette to break up cellular clumps. Take care that vessels do not gets attached to pipette tips. Once per hour with 5 times up and down is in most cases sufficient.
- Spin down the cells at 800 x g for 5 min, remove the supernatant and dissolve the pellets in respective growth medium with growth factors.
- Plate endothelial cells in one PDL/laminin coated well of a 12-well plate and pericytes/smooth muscle cells on two uncoated wells of a 24-well plate at 37 °C with 5% CO2 overnight.
- The next day transfer the supernatant to a new well of the same dish which was also coated with PDL/laminin (but still contains the laminin media till use) for endothelial cells or uncoated for pericyte/vSMCs. Add fresh growth medium with growth factors.
- Change medium every 3-4 days. Niche cells need several days until they begin to proliferate. Don’t split cells until transfer into co-culture hanging inserts. They should be grown at a high density in order to proliferate (Figure 4 shows the initial cell stages).
- Anesthetize 6-8 adult mice according to the appropriate institutional guidelines. Perform cervical dislocation.
- Isolation of astrocytes
Note: An overview of the isolation procedure can be found in Figure 1. The initial steps are the same as for neurosphere isolation, you can see in the video of Babu et al., 2011, how to dissect the hippocampus.- Two weeks prior to co-culture isolate astrocytes from four 4-week old mice.
- Anesthetize adult mice according to the appropriate institutional guidelines. Perform cervical dislocation.
- Spray the head with 70% ethanol to sterilize the area to minimize contamination through fur.
- With sharp scissors decapitate the mouse at the base of the brain stem.
- Cut the skin sagittal along the midline till an arbitrary point between the eyes. Expose the skull free of overlying skin and subcutaneous tissue.
- Cut the skull between eyes by placing each blade of the scissors into each eye cavity. Next, make two lateral cuts at the base of the skull, following a cut through the skull along the sagittal suture.
- Expose the brain by peeling back the skull with the scissors or forceps and remove the brain from the skull with a small spatula.
- Rinse the brain with PBS and transfer to a plastic dish containing PBS.
- Place Petri dish containing the brain under a dissecting microscope at low magnification and position the brain on its ventral surface. Using fine curved forceps remove the olfactory bulbs while holding the brain in position by the cerebellum.
- Rotate the brain onto the dorsal aspect and using a scalpel make a coronal cut through the brain at the level of the optic chiasm.
- Place the caudal portion of the brain in the petri dish and cut along the longitudinal fissure using a scalpel.
- Under the dissection microscope remove the cerebellum and the diencephalon with forceps.
- Remove the hippocampus by sliding with a 27 G needle around the hippocampus formation. Using fine forceps, take of the hippocampus from the surrounding tissue and remove the attached myelin sheets (white matter), which can be found along the medial edge and at the backside of the hippocampus.
- Place the dissected hippocampus into an Eppendorf tube filled with HBSS on ice until all hippocampi are dissected.
- Prepare enzyme mix 1 from neuronal tissue dissociation kit by adding 50 µl of enzyme P and 1,900 µl of buffer X plus 2 µl of β-mercaptoethanol. Vortex and preheat the mixture at 37 °C for 10-15 min before use.
- Remove the HBSS carefully from the hippocampi and mince the tissue in a petri dish using a scalpel blade until no large pieces remain.
- Transfer the minced tissue into a 15 ml Falcon tube with a transfer pipette and add preheated enzyme mix 1.
- Incubate for 15 min at 37 °C with continuous shaking using a tube rotator.
- Prepare enzyme mix 2 from neuronal tissue dissociation kit by adding 40 µl of buffer Y and 20 µl of enzyme A.
- Add enzyme mix 2.
- Triturate 10 times slowly using a medium-sized fire-polished pipette.
- Incubate 10 min at 37 °C.
- Triturate 10 times slowly using a small-sized fire-polished pipette.
- Wash the cells by adding HBSS buffer to a final volume of 10 ml.
- Apply cells in solution to a 40 µm cell strainer, wash the strainer with additional 4 ml HBSS.
- Centrifuge the cells in the 15 ml tube at 300 x g for 5 min.
- Remove the supernatant and resuspend the pellet in the appropriate amount of astrocyte media (see Recipe 5) with 10 ng/ml EGF and 2% FBS.
- Seed onto one well of 12-well plate PDL coated plates and incubate at 37 °C with 5% CO2.
- After 36 h take off the medium and wash the remaining adherent cells thoroughly with pre-warmed PBS and add fresh media with 5 ng/ml EGF and 0.5% FBS.
- Change medium every 3 days, do not passage cells before use in co-culture experiment (see Figure 5 for representative images).
- Two weeks prior to co-culture isolate astrocytes from four 4-week old mice.
- Seeding of the niche co-culture cells into cell inserts
- Two days prior to co-culture dissociate the cells from the different preparations with accutase or trypsin.
Note: We use accutase for astrocyte preparations and trypsin for endothelial and pericyte preparations. - Seed ~5,000 cells per hanging insert in their respective growth medium for the different niche cells cultures (approx. 100 µl per insert, see Recipes 3-5 for media composition), no media is placed in the well below the hanging insert.
- 5-10 h prior to co-culture remove respective growth medium from hanging insert (by aspiration or with pipette) and replace by DMEM with B27 (see Recipe 6) without adding any growth factors.
- Two days prior to co-culture dissociate the cells from the different preparations with accutase or trypsin.
- Isolating hippocampal precursor cells for neurosphere co-culture assay
The isolation procedure for neural precursor cell is according to the protocol of Babu et al. (2011), Walker and Kempermann (2014) with light modifications.- Anesthetize one adult 6 to 8-week-old mouse according to the appropriate institutional guidelines. Perform cervical dislocation.
- Spray the head with 70% ethanol to sterilize the area to minimize contamination through fur.
- With sharp scissors decapitate the mouse at the base of the brain stem.
- Cut the skin sagittal along the midline till an arbitrary point between the eyes. Expose the skull free of overlying skin and subcutaneous tissue.
- Cut the skull between eyes by placing each blade of the scissors into each eye cavity. Next, make two lateral cuts at the base of the skull, following a cut through the skull along the sagittal suture.
- Expose the brain by peeling back the skull with the scissors or forceps and remove the brain from the skull with a small spatula.
- Rinse the brain with PBS and transfer to a plastic dish containing PBS.
- Place petri dish containing the brain under a dissecting microscope at low magnification and position the brain on its ventral surface. Using fine curved forceps remove the olfactory bulbs while holding the brain in position by the cerebellum.
- Rotate the brain onto the dorsal aspect and using a scalpel make a coronal cut through the brain at the level of the optic chiasm.
- For the microdissection of the dentate gyrus (for more details see Walker and Kempermann, 2014) place the caudal portion of the brain in the petri dish and cut along the longitudinal fissure using a scalpel.
- With forceps remove under the dissection microscope the cerebellum and the diencephalon.
- To remove the dentate gyrus, insert the tip of a 27 G needle and slide along the border between the dentate gyrus and Ammon’s horn. Using fine forceps, take off the dentate from the surrounding tissue.
- Place the dissected dentate gyrus in a Petri dish with HBSS on ice.
- Prepare enzyme mix 1 from Neuronal tissue dissociation kit by adding 25 µl of enzyme P and 950 µl of buffer x plus 1 µl of β-mercaptoethanol. Vortex and preheat the mixture at 37 °C for 10-15 min before use.
- Remove the HBSS and mince the tissue using a scalpel blade until no larger pieces remain.
- Transfer the minced tissue from one mouse into a 15 ml Falcon tube and add preheated mix 1.
- Incubate for 15 min at 37 °C with continuous shaking using a tube rotator.
- Prepare enzyme mix 2 from Neuronal tissue dissociation kit by adding 20 µl of buffer Y and 10 µl of enzyme A.
- Add enzyme mix 2.
- Triturate 10 times slowly using a medium-sized fire-polished pipette.
- Incubate 10 min at 37 °C with continuous shaking using a tube rotator.
- Triturate 10 times slowly using a small-sized fire-polished pipette.
- Wash the cells by adding HBSS Buffer to a final volume of 10 ml.
- Apply cells in solution to a 40 µm cell strainer, wash the strainer with additional 4 ml HBSS.
- Centrifuge the cells in the 15 ml tube at 300 x g for 5 min.
- Remove the supernatant and resuspend the pellet in the appropriate amount of DMEM media (see Recipe 6), add 20 ng/ml EGF, 20 ng/ml bFGF and 2 µg/ml heparin.
Note: Depending on how many co-culture conditions are going to be analyzed, titrate the volume of the medium so that enough but not too many cells are seeded per 24-well, otherwise spheres start to fuse. Per 24-well at least 500 µl medium needs to be used in order to have contact to the hanging cell insert.
- Anesthetize one adult 6 to 8-week-old mouse according to the appropriate institutional guidelines. Perform cervical dislocation.
- Hippocampal niche neurosphere co-culture
- Transfer 500 µl of neural cell suspension (from step E26) in DMEM media with growth factors to each well of the 24-well plate.
Note: The number of wells seeded depends on the number of co-culture conditions; in our experiments we seeded neurospheres from one animal into 12 wells of a 24-well plate (using 6 different co-cultures conditions). Do not forget one or two reference wells without niche cells in co-culture, where only stem and progenitor cells are seeded in but only an empty hanging insert with medium is placed on top. The growth of neurospheres in these reference wells can be taken as a baseline to evaluate the influence of the niche co-culture. - Incubate for 30 min at 37 °C with 5% CO2.
- After cells from the isolation have settled down, add hanging inserts with the appropriate cells in DMEM/B27 without growth factors and incubate at 37 °C with 5% CO2 for 10 days. After 10 days take out the hanging inserts and count and measure the spheres using a 10x objective with a scale plate inside on an inverted microscope.
Note: For easier analysis of spheres number draw gridlines on the surface of the 24-well plate or use a transparency with gridline (Figure 6). - For further analysis of differentiation, remove medium containing the spheres from the well and centrifuge at 300 x g for 5 min.
- Resuspend in DMEM medium with B27 but without growth factors and transfer to 24-well plate either with a glass bottom or containing coverslips (both need to be coated with laminin).
- After approximately 7 days at 37 °C with 5% CO2, the differentiation into neurons and astrocytes can be analyzed. Therefore fix cells with 4% paraformaldehyde (pre-warmed to 37 °C) and stain against Map2ab and GFAP (for detail see Walker and Kempermann, 2014).
- Transfer 500 µl of neural cell suspension (from step E26) in DMEM media with growth factors to each well of the 24-well plate.
Representative data
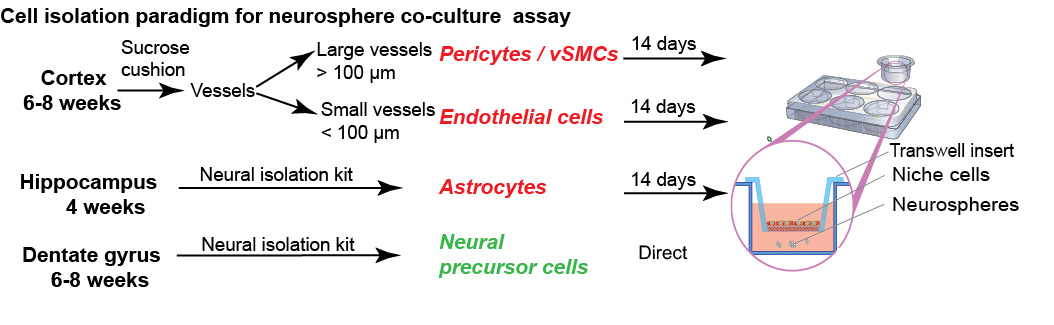
Figure 1. Isolation paradigm for neurosphere co-culture assay. Illustration outlines the primary cell isolation paradigm and the source of tissue, which can be used in this neurosphere co-culture assay. Pericytes/vascular smooth muscle cells (vSMCs) and endothelial cells can be isolated from the cortex, astrocytes were isolated from hippocampus and the dentate gyrus was used for isolation of neurospheres. Niche cells were plated into transwell hanging inserts, while isolated stem cells get seeded below.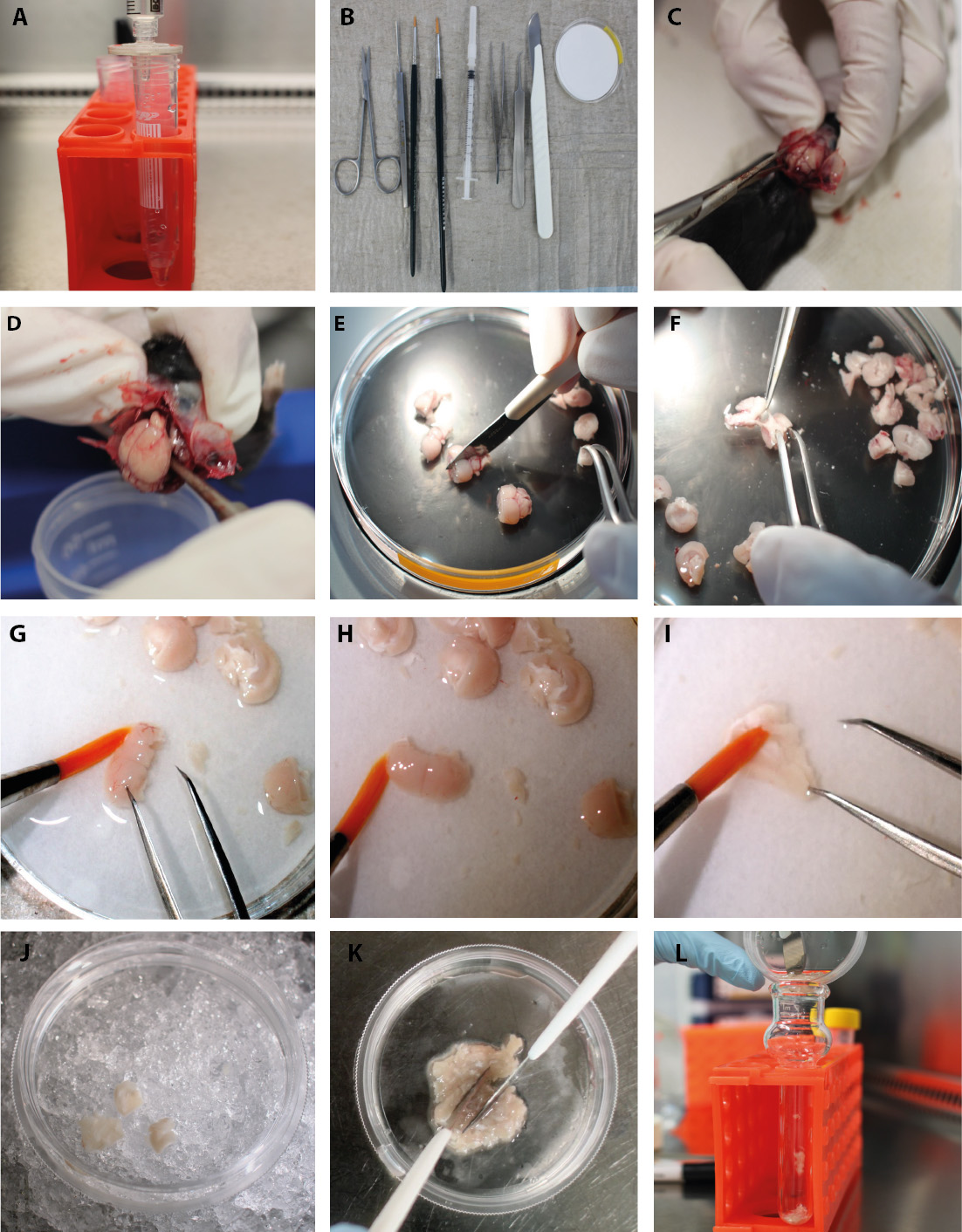
Figure 2. Part 1-Isolation of endothelial cells and pericytes/vSMCs. Step-by-step procedure of the endothelial cell and pericyte/vSMC protocol. A. Sterile filtration of dextran. B. Set of instruments needed for the isolation. C. Sagittal cut through the skull and peel back of the skin and the skull. D. Remove the brain from the skull. E. Coronal cut through the brain with scalpel. F. Take the midbrain off with forceps. G. Remove visible surface blood vessels with a fine brush. H. No blood vessels are visible on the surface after brushing. I. Remove myelin sheets (white matter of corpus callosum) with the brush. J. Transfer the processed cortex to a new petri dish with PBS/FBS on ice. K. Cut the cortex with scalpel blades. L. Transfer the tissue to the douncer.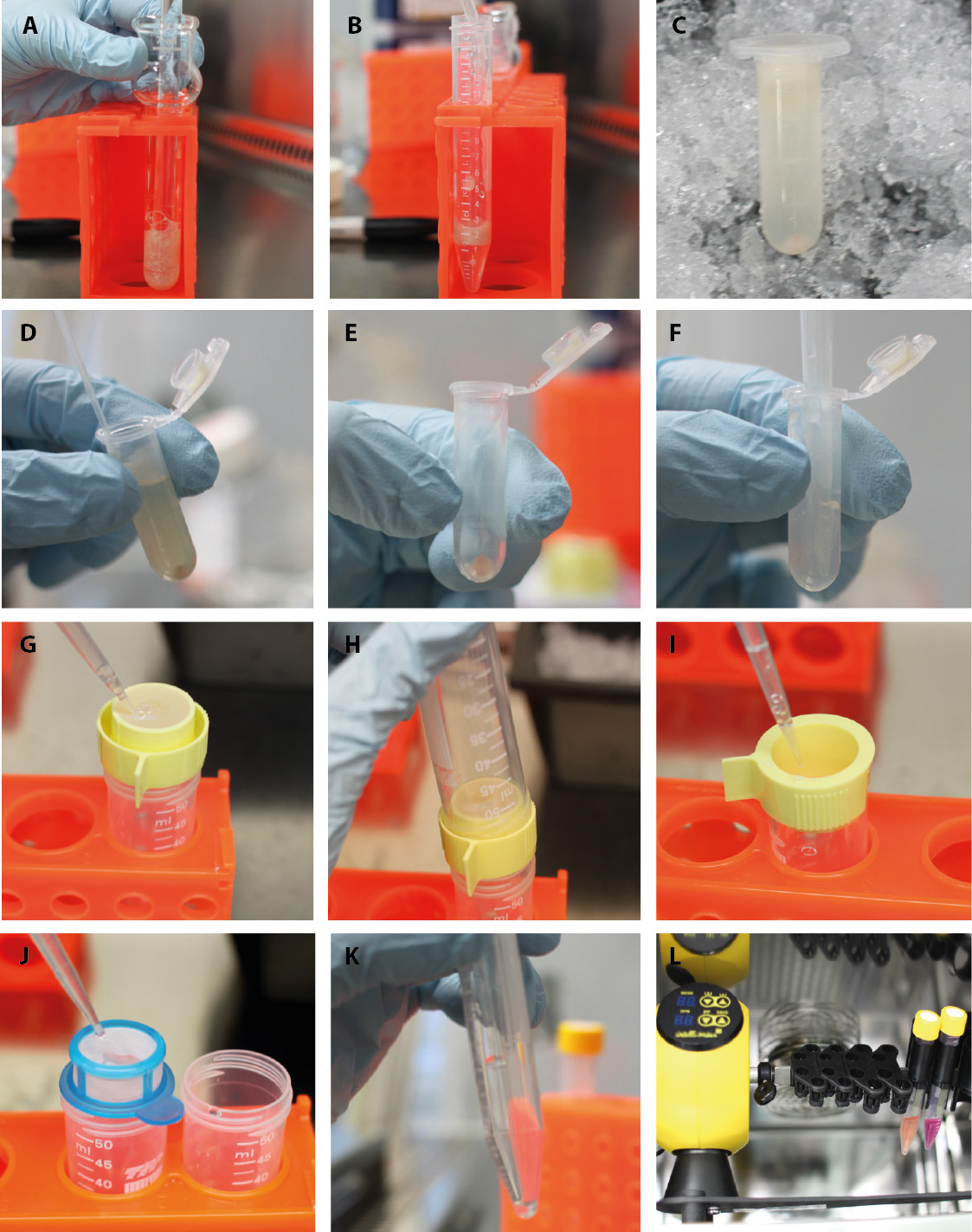
Figure 3. Part 2-Isolation of endothelial cells and pericytes/vSMCs. Second part of the step-by-step procedure of pericyte/vSMC and endothelial cell isolation. A. Homogenize tissue in the douncer with a loose pestle. B. Mix the tissue 1:1 with 30% dextran. C. After centrifugation, a red vessel pellet is observable at the bottom and tissue homogenate floats on top. D. Aspirate carefully the tissue homogenate and the sucrose. E. The clean vessel pellet can be found at the bottom. F. Resuspend the vessel pellet in PBS/FBS. G. Filter vessels in solution through 100 µm nylon strainer, which is turned upside down. H. Take a new tube and flip on top to safely transfer the big vessels into it. I. Rinse the strainer till all vessels are moved back into solution. J. Transfer flow-through from previous step to 40 µm strainer. K. After centrifugation of big and small vessel fraction, pellets can be found. These pellets get now resuspend in collagenase solution for digestion. L. Transfer the tubes with vessel preparations to rotary shaker in the incubator.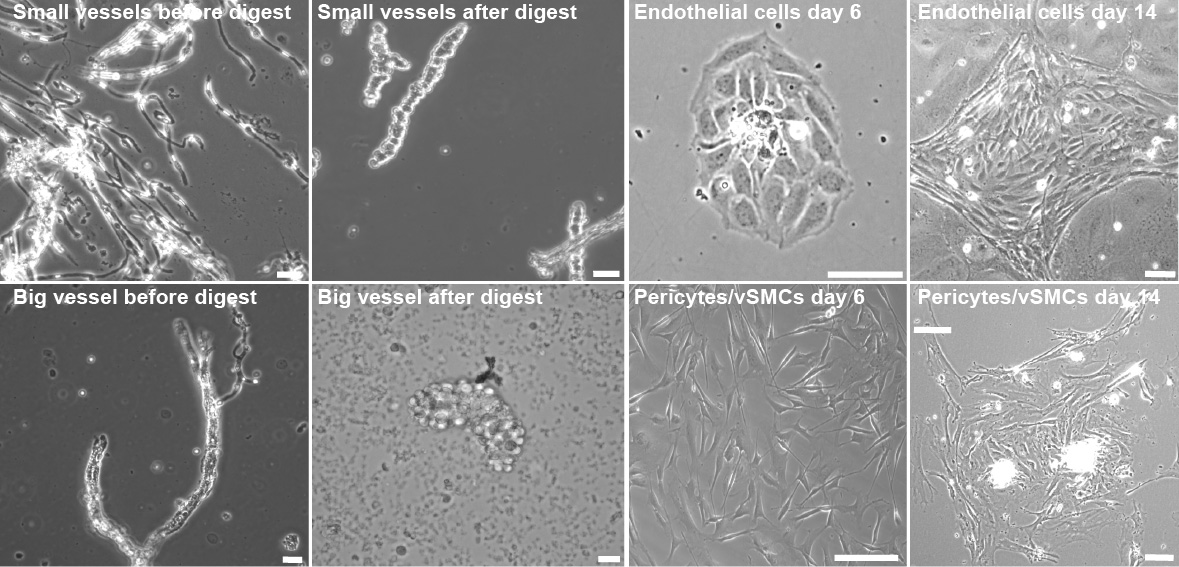
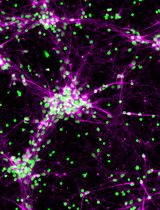
Figure 4. Endothelial cell and pericyte culture after vessel digestion. Representative images of endothelial cells and pericytes/vSMCs during the early time points of preparation. Images taken during the critical steps of endothelial cells (top panel) isolation and pericyte/vSMCs isolation (bottom panel). Scale bar, 10 µm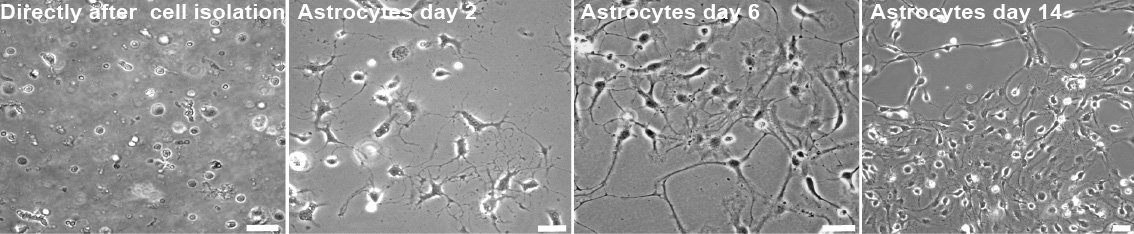
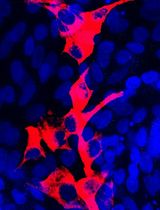
Figure 5. Astrocytes isolation. Representative Images of astrocytes during the early time points of preparation. Images taken during the critical steps of astrocyte isolation. Scale bar, 10 µm
Figure 6. Analysis of neurosphere coculture. Analysis of neurosphere co-culture assay by placing gridlines below. Left, gridlines underneath the 24-well plate. Middle, close-up view of one well. Right, view through the microscope with visible gridlines. Scale bar, 500 µm
Notes
In order to confirm enrichment and sustainability of the different primary niche cell preparations, always seed some cells onto PDL/laminin coated glass slides to evaluate the preparation by immunocytochemistry or take samples for RNA isolation (for details see original publication Ehret et al., 2015).
Recipes
- PBS with 2% FBS
Add 1 ml sterile filtered FBS to 49 ml autoclaved PBS - 30% dextran solution
Dissolve 0.9 g dextran (avg. MW 150,000) in 3 ml HBSS
Dissolve overnight on shaker, filter sterile with tip filters and store at 4 °C for maximal 3 days - Endogrow media (for endothelial cell culture)
MCDB131 media
5% plasma derived platelet poor serum
15 U/ml heparin
ITS premix (5 μg/ml insulin, 5 μg/ml transferrin and 5 ng/ml selenium) (100x stock)
100 U/ml pen/strep (100x stock)
Just before use, add 10 ng/ml EGF, 10 ng/ml FGF-basic and 5 ng/ml VEGF - Pericyte/vSMC media
DMEM with glutamax (100x stock)
10% FBS
0.1 mM non-essential amino acids (100x stock)
100 U/ml Pen/Strep (100x stock)
Just before use, add 10 ng/ml VEGF - Astrocyte media
DMEM with glutamine and HEPES
Glutamax (100x stock)
B27 (50x stock)
Growth factors (EGF and FBS) are added just before use, see steps C27-29 for the different concentrations - Neurosphere media
DMEM without glutamine
B27 (50x stock)
Glutamax (100x stock)
Pen/Strep (100x stock)
Growth factors (EGF and FGF 20 µg/ml each) are added just before use
Acknowledgments
This protocol was used in our previous published study (Ehret et al., 2015). This work was financed by basic institutional grants from the DZNE.
References
- Babu, H., Claasen, J. H., Kannan, S., Runker, A. E., Palmer, T. and Kempermann, G. (2011). A protocol for isolation and enriched monolayer cultivation of neural precursor cells from mouse dentate gyrus. Front Neurosci 5: 89.
- Ehret, F., Vogler, S. and Kempermann, G. (2015). A co-culture model of the hippocampal neurogenic niche reveals differential effects of astrocytes, endothelial cells and pericytes on proliferation and differentiation of adult murine precursor cells. Stem Cell Res 15(3): 514-521.
- Walker, T. L. and Kempermann, G. (2014). One mouse, two cultures: isolation and culture of adult neural stem cells from the two neurogenic zones of individual mice. J Vis Exp(84): e51225.
- Wu, Z., Hofman, F. M. and Zlokovic, B. V. (2003). A simple method for isolation and characterization of mouse brain microvascular endothelial cells. J Neurosci Methods 130(1): 53-63.
Article Information
Copyright
© 2016 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Ehret, F., Vogler, S. and Kempermann, G. (2016). Neurosphere Co-culture Assay. Bio-protocol 6(15): e1883. DOI: 10.21769/BioProtoc.1883.
Category
Stem Cell > Adult stem cell > Neural stem cell
Cell Biology > Cell isolation and culture > Cell differentiation
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.