- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Preparation of Respiratory Syncytial Virus with High or Low Content of Defective Viral Particles and Their Purification from Viral Stocks
Published: Vol 6, Iss 10, May 20, 2016 DOI: 10.21769/BioProtoc.1820 Views: 18783
Reviewed by: Yannick DebingChang Ho LeeDavid Paul
Original research article
The authors used this protocol in:

Sep 2015
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
Respiratory syncytial virus (RSV) belongs to the paramyxovirus family that includes many clinically relevant viruses, such as the human metapneumovirus and measles. RSV infection can cause severe disease in infants, the elderly, and some immunocompromised adults. During RSV replication, a series of truncated forms of the viral genome is generated. These truncated viral genomes are known as defective viral genomes (DVGs) and are generated by many viruses (Lazzarini et al., 1981; Rao and Huang, 1982; Prince et al., 1996; Sun et al., 2015; Tapia et al., 2013). DVGs can restrict the replication of the full-length virus and are the primary natural triggers of the innate immune response to RSV (Sun et al., 2015; Tapia et al., 2013). Here we discuss in detail how to prepare RSV stocks with a high or low content of DVGs, and how to purify defective viral particles containing DVGs from an RSV stock enriched in defective viral particles. These procedures are useful for the preparation of viral stocks and defective viral particles necessary for laboratory research. In brief, the different RSV stocks are produced in HEp2 cells, which are commonly used to amplify this virus in the laboratory. To generate an RSV stock with a high content of DVGs, HEp2 cells are sequentially infected with a high multiplicity of infection (MOI) multiple times followed by purification of the viral particles containing DVGs using gradient centrifugation. The procedure describe here has four parts: 1. Amplification of seed RSV stock with a low DVG content (RSV-LD), 2. Generation of a stock with a high DVG content (RSV-HD), 3. Purification of DVGs by gradient centrifugation, 4. Characterization of purified DVGs.
Keywords: Respiratory syncytial virusMaterials and Reagents
- Sterile polypropylene conical 15 and 50 ml tubes (BD, Falcon®, catalog number: 352070 , or equivalent)
Note: Currently, it is “Corning, Falcon®, catalog number: 352070 ”. - Disposable cell scraper (Thermo Fisher Scientific, catalog number: 08-100-241 )
- Sterile screw-cap microtubes, 2 ml (SARSTEDT AG & Co., catalog number: 15071353 )
- Sterile, aerosol-resistant micropipette tips (1-1,000 μl capacity) (Eppendorf AG or equivalent)
- Cotton-plugged, sterile serological pipettes (1-25 ml capacity) (Eppendorf AG or equivalent)
- Straight-neck polystyrene tissue culture flasks with vented caps, 75 cm2 (Bioexpress, catalog number: T-3001-2 ) and 225 cm2 (BD, FalconTM, catalog number: 353139 )
Note: Currently, it is “Corning, Falcon®, catalog number: 353139”. - Polystyrene tissue culture plates with lids 12 (Greiner Bio-One GmbH, catalog number: 665180 ) and 96 (Greiner Bio-One GmbH, catalog number: 655086 )
- 0.22 μm filter for pipette aid (VWR International, catalog number: 28145-481 )
- Vacuum-driven Stericup® 500 ml Millipore ExpressPLUS 0.22 μm PES (Merck Millipore Corporation, model: SCGPU05RE )
- Ultra-centrifuge tubes
- HEp2 cells (ATCC, catalog number: CCL-23 )
- Human RSV strain A2 (ATCC, catalog number: VR-1540 )
- Mycoplasma removal agent (MP BioMedical, catalog number: 093050044-5 ml )
- Dulbecco’s modified eagle medium (DMEM) (Life Technologies, catalog number: 11995073 )
Note: Currently, it is “Thermo Fisher Scientific, GibcoTM, catalog number: 11995073”. - UltraPureTM EDTA (0.5 M) (Gibco, catalog number: 15575 )
Note: Currently, it is “Thermo Fisher Scientific, InvitrogenTM, catalog number: 15575”. - Trypsin-EDTA, 0.25% (wt/vol) (Thermo Fisher Scientific, GibcoTM, catalog number: 25300054 )
- Fetal bovine serum (FBS), heat-inactivated at 56 °C for 30 min (Thermo Fisher Scientific, catalog number: 10082-147 )
Note: Aliquots should be stored at -20 °C and thawed before use. - Gentamicin reagent solution (Thermo Fisher Scientific, GibcoTM, catalog number: 15750-060 )
- Sodium pyruvate solution (Thermo Fisher Scientific, InvitrogenTM, catalog number: 11360070 )
- L-glutamine (Thermo Fisher Scientific, InvitrogenTM, catalog number: 25030-081 )
- Hanks’ balanced salt solution (HBSS) (Thermo Fisher Scientific, GibcoTM, catalog number: 14025-092 )
- Phosphate-buffered saline (PBS) (pH 7.4) (Thermo Fisher Scientific, GibcoTM, catalog number: 10010056 )
- Ethanol (Thermo Fisher Scientific, DeconTM, catalog number: 64-17-5 )
- Methanol (HPLC) (Thermo Fisher Scientific, Fisher Scientific, catalog number: A452 )
- Crystal violet (Thermo Fisher Scientific, catalog number: C581 )
- Dry ice
- Thermo ScientificTM GeneRulerTM 100 bp Plus DNA Ladder 100 to 3,000 bp (Thermo Fisher Scientific, catalog number: FERSM0322 )
- Electrophoresis, loading dyes, Thermo Scientific, 6x DNA loading dye (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: R0611 )
- UltraPure agarose (Thermo Fisher Scientific, InvitrogenTM, catalog number: 16500-500 )
- Tris-acetate-EDTA (TAE), 10x (Thermo Fisher Scientific, InvitrogenTM, catalog number: 15558026 )
- D-sucrose (Sigma-Aldrich, catalog number: BP220 )
Note: Currently, it is “Thermo Fisher Scientific, catalog number: BP220”. - Gelatin, from bovine skin (Sigma-Aldrich, catalog number: G9391 )
- Superscript® III first-strand synthesis system (Thermo Fisher Scientific, InvitrogenTM, catalog number: 18080-051 )
- Platinum Taq DNA polymerase (Thermo Fisher Scientific, InvitrogenTM, catalog number: 10966018 )
- TRIzol reagent (Thermo Fisher Scientific, AmbionTM, catalog number: 15596018 )
- dNTP set (100 mM) (Thermo Fisher Scientific, InvitrogenTM, catalog number: 10297117 )
- PierceTM coomassie plus (Bradford) protein assay (Thermo Fisher Scientific, catalog number: 23236 )
- RT-PCR primers
- DI1 primer: 5’-CTTAGGTAAGGATATGTAGATTCTACC-3’
- gRSV/DI primer: 5’-CCTCCAAGATTAAAATGATAACTTTAGG-3’
- DI1 primer: 5’-CTTAGGTAAGGATATGTAGATTCTACC-3’
- Regular tissue culture medium (TCM) (see Recipes)
- Infection medium (see Recipes)
- PNE buffer (see Recipes)
- 20% sucrose (see Recipes)
- 0.1% Gelatin in PBS (see Recipes)
- 1% crystal violet stock solution (see Recipes)
- Crystal violet working solution (see Recipes)
Equipment
- Class II biological safety cabinets
- Approved BSL-2 or enhanced BSL-2 laboratory facilities
- Personal protective equipment (PPE)
- Spray bottles for 70% (vol/vol) ethanol
- Micropipettes (1-1,000 μl capacity), multiple channel pipettes (1-200 μl capacity)
- Pipette controller (VWR International)
- Vortex mixer
(Thermo Fisher Scientific)
- Water bath, 37 °C
(Thermo Fisher Scientific)
- Refrigerated table-top centrifuge (for 15 ml and 50 ml conical tubes) (Eppendorf AG, model: 5810R )
- OptimaTM L-90K ultracentrifuge (Beckman Coulter, catalog number: 365670 )
- SW 32 Ti rotor package, swinging bucket (fit for 25 x 89 mm tube) (Beckman Coulter, catalog number: 369694 )
- SW 41 Ti rotor package, swinging bucket (fit for 14 x 89 mm tube) (Beckman Coulter, catalog number: 331362 )
- Gradient Master (BioComp Instruments Inc., catalog number: 107-201M )
- Freezers and refrigerators: -80 °C, -20 °C, and 4 °C
- Tissue culture incubator (37 °C, 5-7% CO2) (Thermo Fisher Scientific)
- Cell counter
- Cell culture microscope (Nikon Corporation or equivalent)
- Microwave
- Glass Erlenmeyer flask (500 ml)
- Agarose gel casting tray (Thermo Fisher Scientific)
- Electrophoresis chamber (Thermo Fisher Scientific)
- Power supply (Thermo Fisher Scientific)
- UV light box (Thermo Fisher Scientific)
- Gel DocTM XR+ gel imaging system (Bio-Rad Laboratories)
- NanoDrop 1000 spectrophotometer (Thermo Fisher Scientific)
- VariokanTM Flash Multimode Reader (Thermo Fisher Scientific)
Procedure
- Preparation of an RSV seed stock with a low content of defective viral particles (RSV-LD)
Note: This step aims to amplify the original virus purchased from ATCC. The goal is to have enough material to then generate an RSV-HD stock.- For initial amplification of the virus, seed 5 x 105/well mycoplasma-free HEp2 cells in TCM in a 6-well plate one night before virus infection.
Note: Cells and viral infections in this protocol are incubated in a tissue culture incubator at 37 °C, 7% CO2 (5% CO2 can also be used). - Next day, wash the cells twice with sterile PBS and infect with RSV at an MOI of 0.01 tissue culture infectious dose (TCID50)/cell. Use the seeded number of cells for MOI calculation. Dilute the virus in infection medium and use a total volume of 200 μl per infection. Incubate at 37 °C in a tissue culture incubator for 2 h.
- Rock the plates once every 15-20 min to maintain an even virus distribution and avoid the cells drying out.
- After 2 h of incubation, add 2 ml infection medium (see Recipes) per well and incubate in a tissue culture incubator.
- Harvest the virus 5 days post-infection by scrapping the cells and collecting them together with the culture supernatants.
- Centrifuge the pooled cells and supernatants for 5 min at 280 x g in a table-top centrifuge at 4 °C and separate the supernatant leaving approx. 200 μl of supernatant in the tube with the pelleted cells.
- Resuspend the cell pellet with the leftover 200 μl supernatant and quick-freeze the mixture in dry ice/ethanol, followed by quick thaw in a 37 °C water bath. Repeat this quick freeze/thaw step at least 3 times and vortex after every time.
- Pool all the freeze-thawed cell debris with the saved supernatant, vortex to mix well, aliquot (500 μl/tube), and quick-freeze the virus in dry ice/ethanol. This is Passage 1 virus (P1).
- Amplify this P1 virus a second time (P2).
- For P2 amplification, seed 3 x 106 HEp2 cells in one T-75 flask and infect with 200 μl P1 virus diluted in 1 ml infection medium, following the same procedure as P1. After 2 h of incubation in a tissue culture incubator add 9 ml of infection medium to each flask.
- Harvest the virus 5 days after infection as described for P1. Aliquot (100 μl/vial) and quick-freeze the virus.
- Titrate the P2 (containing both supernatants and debris) before proceeding with the RSV-HD preparation (for titration method please see TCID50 section below).
Note: Cell death should be noticed 4-5 days post infection (Figure 1). If earlier cell death is noticed, reduce the amount of P1 virus. If no cell death is noticed post 5 days infection, passage P2 another time following steps A10-12.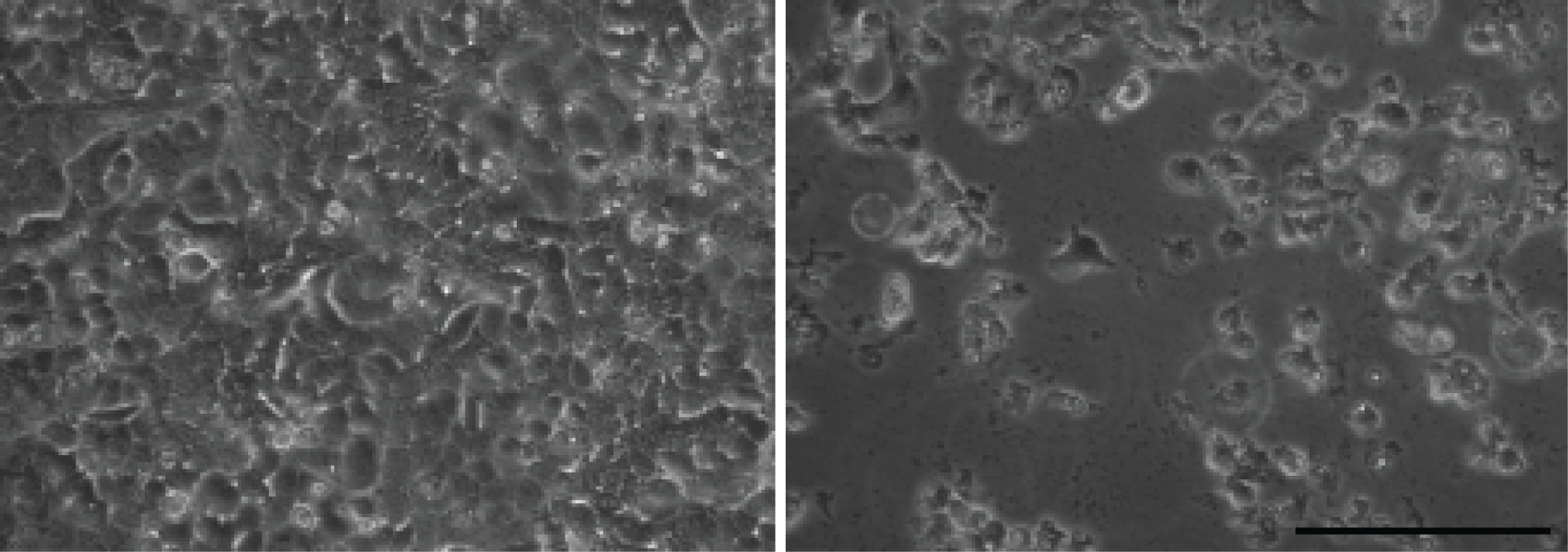
Figure 1. Cell death during RSV infection. HEp2 cells were infected with RSV at MOI of 0.1. The images were taken 4 days post infection. Left: mock infection. Right: RSV-LD infection. Scale bar: 0.1 mm
- For initial amplification of the virus, seed 5 x 105/well mycoplasma-free HEp2 cells in TCM in a 6-well plate one night before virus infection.
- Preparation and concentration of an RSV stock with a high content of defective viral particles (RSV-HD).
Note: This step aims to enrich DVGs in the RSV stock. The goal is to have enough DVG-containing defective viral particles to purify from the viral stock. The standard way to promote DVG production during viral infection is to sequentially passage the virus stock (generated from section A in vitro using a high MOI.- To prepare an RSV-HD seed stock, infect HEp2 cells with RSV-LD P2 at a moi of 4 TCID50/cell for two passages to generate RSV-HD P1 and P2. Use the same procedure as for LD preparation. Specifically, T75 flasks are used for RSV-HD P1 and P2 amplification with an inoculum of 1 ml/flask. Additional 9 ml of infection medium should be added after 2 h of incubation. Calculate the viral titers using the TCID50 assay (see TCID50 section).
- Harvest viruses 2 days post-infection or when more than half of the cells are dead. Quick freeze/thaw the cell pellet at least 3 times as described in steps A6-7.
- Collect the supernatants and discard the cell debris. Aliquot (500 μl/vial) and quick-freeze the viruses.
- To generate an RSV-HD working stock, seed 107 HEp2 cells in one T-225 flask and infect with RSV-HD P2 at a moi of 10 TCID50/cell. Use an inoculum of 4 ml per T-225 flask and follow the infection procedure described above. Add 21 ml of infection medium after 2 h incubation of viruses and put it back in the tissue culture incubator.
- Two days later, or when more than half of the cells have detached from the plate, scrape the cells and collect them together with the culture supernatant to harvest the virus.
- Centrifuge for 5 min at 280 x g in a table-top centrifuge at 4 °C and collect the supernatant.
- Return 2-3 ml of the supernatant to each pellet, and put the rest of the supernatants on ice. Suspend the pellet, vortex, and quick freeze/thaw the mixtures at least 3-4 times as in steps A6-7.
- Centrifuge for 5 min at 280 x g in a table-top centrifuge, the goal is to eliminate debris. Take the supernatants and combine them with the rest of the supernatants.
Caution: Discard the debris at this step. - Ultra-centrifuge the supernatants for 2.5 h at 59,000 x g at 4 °C using an SW 32 Ti Rotor and Ultra-ClearTM 25 x 89 mm centrifuge tube to concentrate the RSV-HD working stock.
- Lightly wash with 1 ml of HBSS twice without disturbing the pellet.
- Suspend the pellet in a total 200 μl of infection medium / T225 flask.
- Aliquot (200 μl /vial) and quick-freeze the viruses in dry ice/ethanol for storage.
- In order to obtain enough defective viral particles, we usually infect 48 T225-flasks, totaling 9 ml of ultra-centrifuged RSV-HD (less virus may be sufficient but the yield decreases).
- To prepare an RSV-HD seed stock, infect HEp2 cells with RSV-LD P2 at a moi of 4 TCID50/cell for two passages to generate RSV-HD P1 and P2. Use the same procedure as for LD preparation. Specifically, T75 flasks are used for RSV-HD P1 and P2 amplification with an inoculum of 1 ml/flask. Additional 9 ml of infection medium should be added after 2 h of incubation. Calculate the viral titers using the TCID50 assay (see TCID50 section).
- Purification of RSV defective viral particles
Note: This step aims to isolate the defective viral particles containing DVGs from the infectious viral particles with full-length genome. Since defective viral particles are much smaller and less dense compared to standard viral particles, the lighter (in our case is the top) fraction contains the most DVGs.- Make 100 ml of PNE buffer fresh in the biosafety hood, shake and mix well.
- Prepare 20% and 60% sucrose solutions using PNE buffer, shake and mix well.
- Set out 6 tubes of Ultra-ClearTM 14 x 89 mm centrifuge tubes.
- Fill each tube with 5 ml of 20% sucrose solution. Carefully and slowly pipette 5 ml 60% sucrose solution starting at the bottom of the tube and slowly lift up the 20% sucrose layer.
Caution: Slow action is critical. A sharp separation between 20% and 60% sucrose solutions should be observed. - Carefully load the tubes containing sucrose solution on the Gradient Master to generate 20%-60% sucrose gradient. Do this in the biosafety hood to maintain sterility.
- Once finished, carefully layer 1.5 ml of ultra-centrifuged RSV-HD (prepared in step B) on top of each sucrose gradient. Use a P1000 micropipette to place the virus. The tubes should be filled up to 2-3 mm from the edge to avoid them collapsing during ultracentrifugation.
Caution: Do not disturb the sucrose gradient when adding the virus. - Ultra-centrifuge the gradients layered with virus for 2 h at 116,000 x g at 4 °C using an SW 41 Ti Rotor and Ultra-ClearTM 14 x 89 mm centrifuge tubes.
- Carefully remove the top pink layer once finished the centrifuge.
Note: you should be able to observe several cloudy layers. Extract the cloudy layers from top to bottom using a P1000 micropipette. Transfer and combine the same fraction from all 6 tubes to a separate clean Ultra-Clear 14 x 89 mm centrifuge tube. In total, 3 layers are extracted from RSV-HD, named Fraction 1, 2, 3 from top to bottom (Figure 2).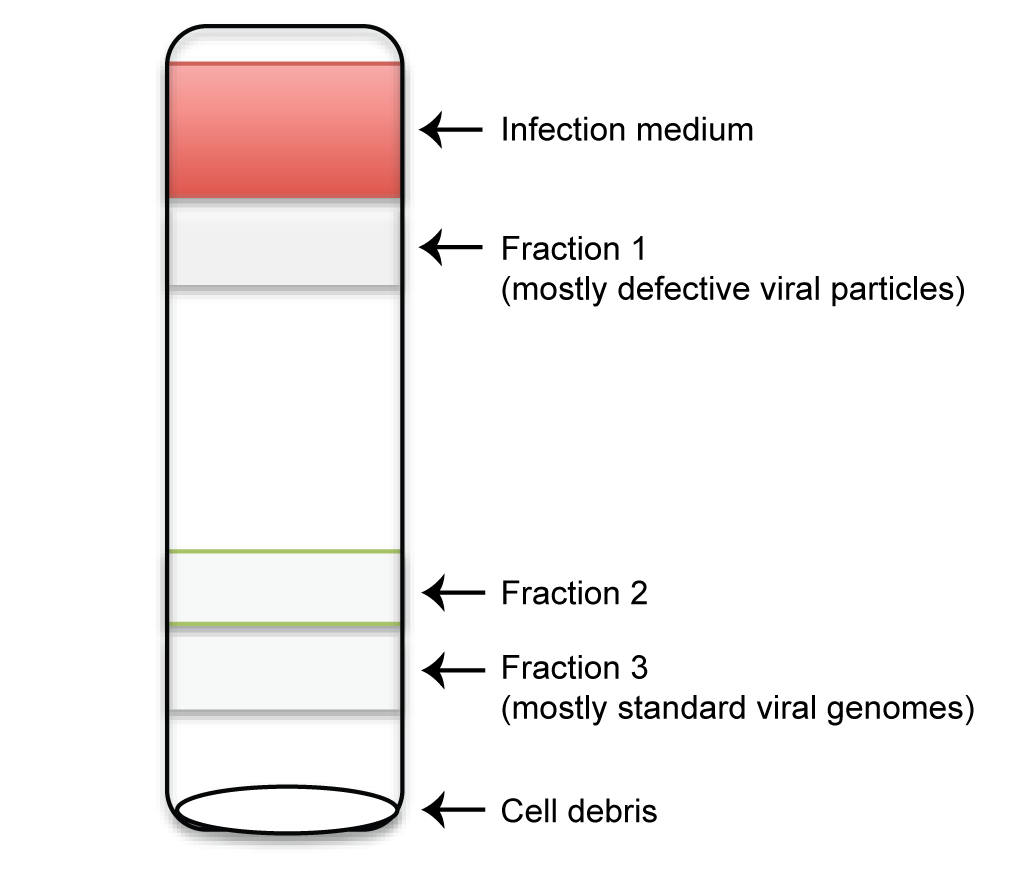
Figure 2. Schematic diagram of defective viral particles purified through sucrose gradient - Fill up each tube to 2-3 mm from the top with PBS and ultra-centrifuge for 2 h at 4 °C at 106,000 x g using SW 41 Ti Rotor.
- Suspend the pellet from each fraction in 1 ml of 0.1% Gelatin in PBS, aliquot 100 μl per vial, snap freeze, and store at -80 °C.
- Make 100 ml of PNE buffer fresh in the biosafety hood, shake and mix well.
- Virus Titration by TCID50 (infectious viral titer)
Note: This step aims to quantify the infectious viral particles contained in each of the purified fractions based on their ability to replicate in permissive cells.- The day before titration prepare 96 well plates with HEp2 cells. Seed the plate with 2 x 104 cells / well in TCM (100 μl /well of a solution of 2 x 106 cells/ 10 ml TCM). One plate will fit four test samples. Prepare as many plates as needed.
- The day of the titration the cells should be 80-90% confluent.
- In a separate 96 well plate, prepare 1/10 serial dilutions of fractions 1, 2, and 3 in triplicates in infection media. To do this add 90 μl of infection media to all wells in the plate. Add 10 μl of each fraction to each well on the first row. Prepare triplicates for each fraction. Leave three rows for media alone, and include a positive control with a virus of known titer. Use a multichannel pipette to mix the virus with the infection media in the first row and transfer 10 μl to the next row. Change tips, mix and transfer 10 μl to the next row, continue until the last row. Make sure to mix the virus well and change the tips for every dilution.
- Remove the TCM of the plate containing the cells and wash the monolayer twice with 100 μl of serum free media.
- Using a multichannel pipette, transfer 25 μl/well of the virus dilutions to the cells and incubate for 2 h at 37 °C in a tissue culture incubator. Start transferring from the bottom of the plate (higher dilution) to avoid carrying over virus from the higher concentrations.
- Add 75 μl of infection media per well and incubate for 4-5 days in a tissue culture incubator. Be careful not to contaminate the media with virus from the plate.
- To determine the viral titer, discard the media in a glass tray containing 10% bleach.
- Add 100 μl of crystal violet working solution to the wells. Be careful NOT to touch the bottom of the wells.
- Wait for 15-30 min.
- Wash the plate by submerging it upside down in water several times to eliminate the excess of crystal violet.
- Let it dry at RT.
- Score the titer by determining the last dilution with positive CPE (Figure 3). Score the number of positive wells for that last dilution (number of positive out of three). From this score calculate the TCID50/25 μl using the following formula, where “x” correspond to the dilution:
“+ + +” 10x TCID50 = 10X+0.7/25 μl
“+ + - ” 10x TCID50 = 10X+0.4/25 μl
“+ - - ” 10x TCID50 = 10X-0.1/25 μl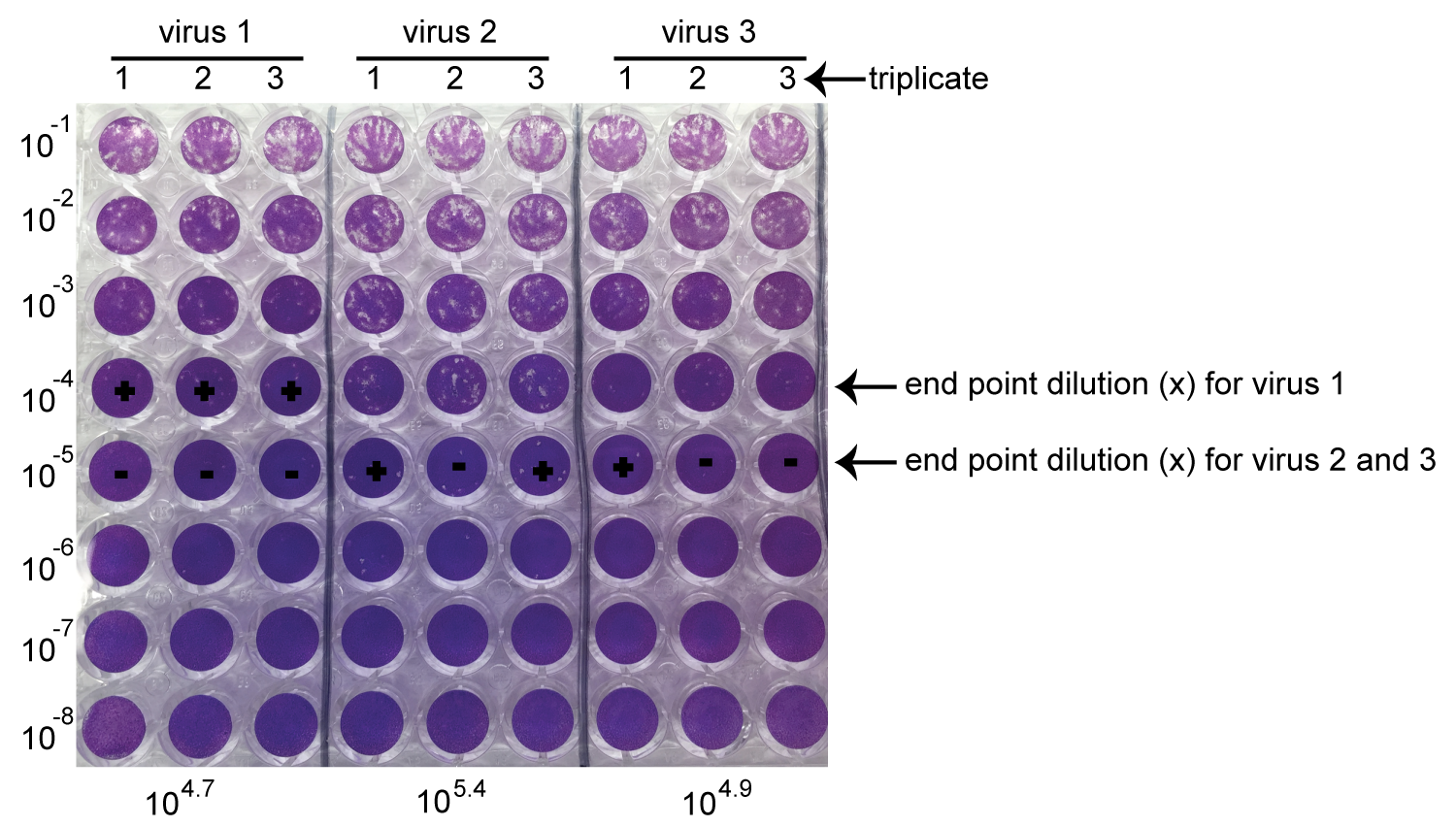
Figure 3. Crystal violet staining of TCID50. The titers from three different viruses were determined by TCID50 as illustrated above. 10-4 and 10-5 were the last dilutions with positive CPE for designated virus samples, which were the “x”. “+” stands for the positive CPE observed. “-” indicates no CPE observed. Based on the equation above, calculate the final titer of each virus as shown at the bottom of this figure.
- The day before titration prepare 96 well plates with HEp2 cells. Seed the plate with 2 x 104 cells / well in TCM (100 μl /well of a solution of 2 x 106 cells/ 10 ml TCM). One plate will fit four test samples. Prepare as many plates as needed.
- Estimation of total amount of viral particles (total virus)
Note: This step aims to estimate the quantity of total viral particles present in each of the purified fractions. We use total amount of protein in each fraction measured by Bradford assay as an estimate of total viral particles.- Dilute 5 μl of Fraction 1, 2, and 3 into 20 μl of dH2O separately → 1:5 dilution.
- Prepare the BSA standard (provided in the kit) using serial dilutions as shown in Table1.
Table 1. BSA serial dilution for standard curve from 2 mg BSA stock
Std number Volume of diluent
(μl)Volume of stock or sample (μl)BSA conc.
mg/ml1 25 75 (BSA: 2 mg/ml) 1.500 2 65 65 (BSA: 2 mg/ml) 1.000 3 35 35 of Std 1 0.750 4 65 65 of Std 2 0.500 5 65 65 of Std 4 0.250 6 65 65 of Std 5 0.125 7 80 20 of Std 6 0.025 - Prepare 15 ml of Coomassie blue reagent (provided in the kit) per 96 wells plate and equilibrate it to room temperate before use.
- Add 150 μl of Coomassie blue reagent per well to the 96 well plate.
- Add 5 μl of the BSA standard and the diluted Fractions 1, 2, and 3.
- Mix by tapping the edge of the plate.
- Incubate the plate for 15 min at room temperature.
- Read O.D at 595 nm using a VariokanTM Flash Multimode Reader or equivalent.
- Based on the standard curve, calculate the total protein concentration of Fractions 1, 2, and 3.
- Dilute 5 μl of Fraction 1, 2, and 3 into 20 μl of dH2O separately → 1:5 dilution.
- Infectivity/Total viral protein ratio calculation
Note: This step aims to determine which fraction contains most of the defective viral particles with the least standard infectious viral particles.- The ratio between Infectivity titer (I) and Total viral protein (T) equals to TCID50 per 25 μl / total viral protein per 25 μl.
- Select the Fraction with the lowest I/T to further validate the defective viral genome content.
- The ratio between Infectivity titer (I) and Total viral protein (T) equals to TCID50 per 25 μl / total viral protein per 25 μl.
- Validation of purified RSV defective viral particles by PCR
Note: This step aims to confirm that the fraction selected in section F is enriched in DVGs. For this purpose, permissive cells are incubated with each purified fraction and DVGs are identified using a specific RT-PCR assay (DVG-RT-PCR). For detailed illustration of the DVG RT-PCR assay please refer to Sun et al. (2015), Figure S1.- HEp2 cells are supplemented with either PBS or 1, 10, 20 μl of the selected Fraction. In a separate well infect with RSV-HD working stock of MOI of 1 as a control.
- Harvest the infected cells 10 h post infection by adding TRIzol. Extract RNA from TRIzol following the manufacturer’s protocol. Quantify the RNA concentration via Nano-drop and use 1 μg of RNA for reverse transcription (RT).
- Mix the following reagents per sample in one tube: 1 μl of the DI1 primer (50 uM), 1 μl of dNTPs (10 mM), 1 μg of RNA, nuclease-free H2O (add up to 10 μl).
- Incubate 10 min at 65 °C.
- Prepare the RT reaction mix in tubes on ice using SuperScript III reverse transcriptase First-Strand System as follows (per sample): 5x RT buffer 2 μl, 25 mM MgCl2 4 μl, 0.1 M DTT 2 μl, RNase OUT 1 μl, SS III 1 μl.
- After incubation, add 10 μl of RT mix to each sample. Mix well.
- Incubate at 50 °C for 50 min.
- Heat at 85 °C for 5 min.
- Add 1 μl of RNase H per sample (from SuperScript III reverse transcriptase kit) and incubate at 37 °C for 20 min.
- cDNA can be stored at -20 °C or directly used for PCR.
- Spin down the cDNA and prepare the DI-PCR mix as below:
Reagent Volume (μl) Water 13.75 Buffer (10x) 2.5 MgSO4 (50 mM) 2.5 dNTPs (10 mM) 2.0 RSV DI1 primer (10 μM) 1.0 DI gSeV Primer (10 μM) 1.0 Taq polymerase (5 units/μl) 0.25 cDNA 2.0 Total 25 - Run PCR Program in a thermocycler.
Program Temperature (°C) Time Denaturation 95 10 min Hold Denaturation 95 30 sec 33-35 cycles Annealing 55 30 sec Extension 72 90 sec Final extension 72 5 min Hold 4 ∞ Hold - Add 4 μl of the loading buffer to each sample.
- Load the samples and 100 bp Plus DNA Ladder in the 1.5% agarose gel.
- Observe the defective viral genome bands as shown in Figure 4.
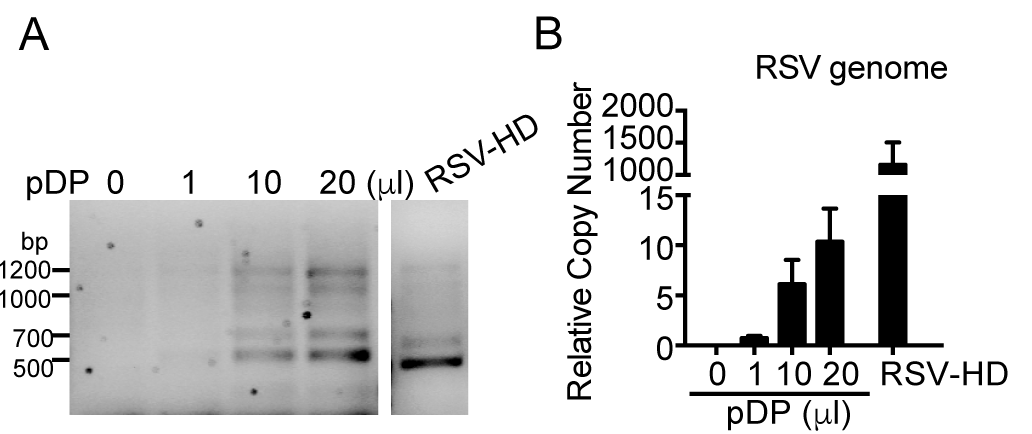
Figure 4. Content of defective viral genomes and standard genomes in Fraction 1. HEp2 cells were incubated with infection medium and supplemented with 0, 1, 10, or 20 μl of purified defective viral particles (pDP) (Fraction 1). Cells were infected with RSV-HD at an MOI of 1, serving as a positive control. Samples were harvested at 10 h post infection. RNA was extracted followed by DVG-RT-PCR to detect DVGs (A) and genome qPCR to quantify the full-length viral genome content (B). The bands observed during pDP supplementation mirrored the pattern in RSV-HD infection. Little viral replication of standard virus was detected during pDP supplementation, demonstrating that this fraction is enriched in DVGs (but not genome). The four major bands were further confirmed by sequencing [see Sun et al. (2015), Figure S2]. For details on the RSV genome qPCR, please refer to Tapia et al. (2013).
- HEp2 cells are supplemented with either PBS or 1, 10, 20 μl of the selected Fraction. In a separate well infect with RSV-HD working stock of MOI of 1 as a control.
Recipes
- Regular tissue culture medium (TCM)
500 ml DMEM supplemented with:
10% fetal bovine serum
1 mM sodium pyruvate (final concentration)
2 mM L-Glutamine (final concentration)
50 mg/ml gentamicin (final concentration)
Pass the solution through a 0.22 μm filter - Infection medium
500 ml DMEM supplemented with:
2% fetal bovine serum (final concentration)
1 mM sodium pyruvate (final concentration)
2 mM L-Glutamine (final concentration)
50 mg/ml gentamicin (final concentration)
Pass the solution through a 0.22 μm filter - PNE buffer
50 ml PBS supplemented with 200 μl of 0.5 M EDTA - 20% sucrose
10 g sucrose dissolved in a final volume of 50 ml of PNE buffer
60% sucrose
30 g sucrose dissolved in a final volume of 50 ml of PNE buffer - 0.1% Gelatin in PBS
Add 500 mg gelatin power to a total volume of in 500 ml dH2O, shake until fully dissolved.
Pass the solution through a 0.22 μm filter
Maintain sterile - 1% crystal violet stock solution (100 ml)
1 g crystal violet
20 ml 100% ethanol
80 ml dH2O - Crystal violet working stock
40 ml 1% crystal violet solution
80 ml methanol
180 ml dH2O
Acknowledgments
This work was supported by grant AI083284 from the National Institute of Health (CBL). The protocol described herein was based on the following paper: Sun et al. (2015).
References
- Lazzarini, R. A., Keene, J. D. and Schubert, M. (1981). The Origins of Defective Interfering Particles of the Negative-Strand Rna Viruses. Cell 26(2): 145-154.
- Lopez, C. B. (2014). Defective Viral Genomes: Critical Danger Signals of Viral Infections. Journal of Virology 88(16): 8720-8723.
- Prince, A. M., HuimaByron, T., Parker, T. S. and Levine, D. M. (1996). Visualization of hepatitis C virions and putative defective interfering particles isolated from low-density lipoproteins. Journal of Viral Hepatitis 3(1): 11-17.
- Rao, D. D. and Huang, A. S. (1982). Interference among Defective Interfering Particles of Vesicular Stomatitis-Virus. Journal of Virology 41(1): 210-221.
- Sun, Y. and López, C. B. (2016). Respiratory syncytial virus infection in mice and detection of viral genomes in the lung using RT-qPCR. Bio-protocol 6(9): e1819.
- Sun, Y., Jain, D., Koziol-White, C. J., Genoyer, E., Gilbert, M., Tapia, K., Panettieri, R. A., Hodinka, R. L. and Lopez, C. B. (2015). Immunostimulatory Defective Viral Genomes from Respiratory Syncytial Virus Promote a Strong Innate Antiviral Response during Infection in Mice and Humans. Plos Pathogens 11(9).
- Tapia, K., Kim, W. K., Sun, Y., Mercado-Lopez, X., Dunay, E., Wise, M., Adu, M. and Lopez, C. B. (2013). Defective viral genomes arising in vivo provide critical danger signals for the triggering of lung antiviral immunity. Plos Pathogens 9(10).
- Yount, J. S., Kraus, T. A., Horvath, C. M., Moran, T. M. and Lopez, C. B. (2006). A novel role for viral-defective interfering particles in enhancing dendritic cell maturation. Journal of Immunology 177(7): 4503-4513.
Article Information
Copyright
© 2016 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Sun, Y. and López, C. B. (2016). Preparation of Respiratory Syncytial Virus with High or Low Content of Defective Viral Particles and Their Purification from Viral Stocks. Bio-protocol 6(10): e1820. DOI: 10.21769/BioProtoc.1820.
- Sun, Y., Jain, D., Koziol-White, C. J., Genoyer, E., Gilbert, M., Tapia, K., Panettieri, R. A., Hodinka, R. L. and Lopez, C. B. (2015). Immunostimulatory Defective Viral Genomes from Respiratory Syncytial Virus Promote a Strong Innate Antiviral Response during Infection in Mice and Humans. Plos Pathogens 11(9).
Category
Microbiology > Microbial biochemistry > Protein > Isolation and purification
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.