- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Affymetrix Genome-wide Human SNP Assay
Published: Vol 6, Iss 10, May 20, 2016 DOI: 10.21769/BioProtoc.1806 Views: 8585
Reviewed by: Francesco Dal GrandeIrit AdiniAnonymous reviewer(s)
Original research article
The authors used this protocol in:

May 2015
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
To assess genomic variation, it is possible to identify the single nucleotide polymorphisms (SNP) which an individual carries. Using the Affymetrix Genome-wide Human SNP Assay, it is possible to assess 906,600 SNPs on a single array. This protocol, the next iteration of the GeneChip Mapping 500K array set, is based directly on the manufacturers’ protocol and shows steps which are highly similar to that which is found here: http://media.affymetrix.com/support/downloads/manuals/genomewidesnp6_manual.pdf.
Keywords: Single nucleotide polymorphismsMaterials and Reagents
- 96-well V-bottom plates, elution catch (Greiner Bio-One GmbH, catalog number: 651101 )
- Greiner UV star transparent, 96-well (Greiner Bio-One GmbH, catalog number: 82050-788 )
Note: Currently, it is “VWR International, catalog number: 82050-788”. - Multiplate 96-well unskirted PCR plate (Bio-Rad Laboratories, catalog number: MLP-9601 )
- Multiscreen deep well plate (Merck Millipore Corporation, catalog number: MDRLN0410 )
- 2.4 ml deep well storage plate/pooling plate (Greiner Bio-One GmbH, catalog number: 780280 )
- 100 ml solution basin (VWR International, LabcorTM, catalog number: 730-014 )
- 55 ml solution basin (VWR International, LabcorTM, catalog number: 730-004 )
- 15 ml centrifuge tubes (VWR International, catalog number: 20171-020 )
- 50 ml centrifuge tubes (VWR International, catalog number: 21008-178 )
- 2.0 ml Eppendorf tubes (VWR International, catalog number: 20901-540 )
- 0.2 ml 12-strip tubes (VWR International, catalog number: 21008-940 )
- Lab tape
- Tough-Spots®
- Microseal ‘B’ adhesive seal (Bio-Rad Laboratories, model: MSB1001 )
- MicroAmp® clear adhesive film applied (Biosystems, catalog number: 4306311 )
Note: Currently, it is “Thermo Fisher Scientific, Applied BiosystemsTM, catalog number: 4306311”. - Kimwipes (source as desired)
- Pipet tips as appropriate for pipettes (source as desired)
- AccuGENE® water, molecular biology-grade (Affymetrix, catalog number: 71786 )
- GenElute Mammalian Genomic DNA Miniprep (Sigma-Aldrich, catalog number: G1N70 )
- Affymetrix SNP 6 Core Reagent Kit, 100 reactions (Affymetrix, catalog number: 901706 )
- 10x Sty I buffer/NE buffer 3 (New England Biolabs)
- T4 DNA Ligase (400 U/μl) (New England Biolabs)
- T4 DNA Ligase Buffer (10x) (New England Biolabs)
- Adaptor, sty (50 μM)
- PCR Primer 002 (100 μM)
- 10x Nsp I buffer/NE buffer 2 (New England Biolabs)
- NspI (10 U/μl) (New England Biolabs)
- Adaptor, nsp (50 μM)
- Elution buffer (Buffer EB)
- Fragmentation buffer (10x)
- Fragmentation reagent (DNase I)
- DNA Labeling reagent
- Terminal deoxynucleotidyl transferase (TdT) (30 U/μl)
- Terminal deoxynucleotidyl transferase buffer (TdT buffer) (5x)
- Oligo control reagent (OCR), 0100
- 10 mg/ml BSA (100x)
- Hybridization Master Mix
- 10x Sty I buffer/NE buffer 3 (New England Biolabs)
- DNA marker (Bionexus, catalog number: BN2050 )
- 2% TBE gel (Thermo Fisher Scientific, InvitrogenTM, catalog number: G8008-02 )
- Gel loading solution (Faster Better Media, catalog number: SB5N-8 )
- Ethanol (Sigma-Aldrich, catalog number: 459844 )
- 50 ml magnetic beads (AMPure, catalog number: A63881 )
Note: It is named “Agencourt AMPure XP-PCR Purification” on Beckman Coulter website. - 4% TBE gel (Thermo Fisher Scientific, InvitrogenTM, catalog number: G8008-04 )
- MES hydrate (Sigma-Aldrich, catalog number: M8250 )
- MES sodium salt (Sigma-Aldrich, catalog number: M3885 )
Note: All catalog numbers are listed in the Affymetrix datasheet (http://media.affymetrix.com/support/downloads/manuals/genomewidesnp6_manual.pdf). - Clontech TITANIUMTM DNA Amplification Kit (Clontech, catalog number: 639240 ) (see Recipes)
- 12x MES Stock Solution (see Recipes)
Equipment
- Applied Biosystems units
- 2720 Thermal Cycler
- GeneAmp® PCR System 9700
- GeneAmp® PCR System 9700 (silver block or gold-plated silver block)
- 2720 Thermal Cycler
- 48 Genome-Wide Human SNP Array 6.0 (one array per sample)
- GeneChip® Hybridization Oven 640
- Jitterbug
- Spectrophotometer plate reader
- Vacuum Manifold (Merck Millipore Corporation)
- Vortexer
- Cooler (chilled to -20 °C)
- Cooling chamber (chilled to 4 °C) (do not freeze)
- Cooling chamber (double, chilled to 4 °C) (do not freeze)
- Ice bucket, filled with ice
- Marker, fine point, permanent
- Microcentrifuge
- Pipet, 12-channel P1200
- Pipet, 12-channel P200
- Pipet, 12-channel P20 (accurate to within ± 5%)
- Pipet, serological
- Pipet, single channel P1000
- Pipet, single channel P200
- Plate centrifuge with deep-well capacity (54 mmH x 160 g)
- Optical plates
- Centrifuge plate holders
Notes:- Use only the PCR plate, adhesive film and thermal cyclers listed here.
- All relevant equipment numbers are listed in the Affymetrix datasheet.
- Use only the PCR plate, adhesive film and thermal cyclers listed here.
Procedure
Genomic DNA Plate Preparation
- Extract DNA from your cells of interest using an extraction protocol which yields high quality, contaminant-free DNA free from inhibitors (e.g., GenElute Mammalian Genomic DNA Miniprep).
Note: DNA should be around 1.8 for 260/280 and 2.0 for 260/230 ratios when examining the optical density using a spectrophotometer. - Thoroughly mix the genomic DNA by vortexing at high speed for 3 sec.
- Determine the concentration of each sample (e.g., Nanodrop).
- Dilute each sample to 50 ng/μl using reduced EDTA TE buffer.
- Thoroughly mix the diluted DNA by vortexing at high speed for 3 sec.
- Vortex the plate of genomic DNA at high speed for 10 sec, then spin down at 560 x g for 30 sec.
- Aliquot 5 μl of each DNA to the corresponding wells of two 96-well reaction plates. 5 μl of the 50 ng/μl working stock is equivalent to 250 ng genomic DNA per well. Two replicates of each sample are required for this protocol: One for Nsp and one for processing Sty.
- Seal each plate with adhesive film.
Stage 1: Sty Restriction Enzyme Digestion
During this stage, the genomic DNA is digested by the StyI restriction enzyme.
- Thaw/place the following reagents on ice:
- NE Buffer 3
- BSA
- Genomic DNA (if frozen)
- AccuGENE water
- NE Buffer 3
- Label the following tubes, then place in the cooling chamber:
- One strip of 12 tubes labeled “Dig”.
- One 2.0 ml Eppendorf tube labeled “Dig MM”.
- One strip of 12 tubes labeled “Dig”.
- Prepare the plate with genomic DNA as follows:
- Vortex the center of the plate at high speed for 3 sec.
- Spin down the plate at 560 x g for 30 sec.
- Place back in the cooling chamber on ice.
- Vortex the center of the plate at high speed for 3 sec.
- Prepare the reagents (except for the enzyme) as follows:
- Vortex 3 times, 1 sec each time.
- Pulse spin for 3 sec.
- Place in the cooling chamber.
- Vortex 3 times, 1 sec each time.
- Preheat the Thermal Cycler lid, however leave the block at room temperature.
Note: Leave the STYI enzyme at -20 °C until ready to use. - Prepare the Sty digestion Master Mix by adding the volumes of the following reagents to the 2.0 ml Eppendorf tube as in Table 1:
- AccuGENE water
- NE Buffer 3
- BSA
- AccuGENE water
- Remove the StyI enzyme from the freezer and immediately place in a cooler.
- Pulse spin the enzyme for 3 sec.
- Immediately add the enzyme to the master mix, then place remaining enzyme back in the cooler.
- Vortex the master mix at high speed 3 times, 1 sec each time.
- Pulse spin for 3 sec.
- Place in the cooling chamber.
- Return any remaining enzyme to the freezer.
- Proceed immediately to Add Sty Digestion Master Mix to Samples.
Table 1. StyI Digestion Master MixReagent 1 Sample 48 Samples (15% Extra) AccuGENE® water 11.55 μl 637.6 μl NE Buffer 3 (10x) 2 μl 110.4 μl BSA (100x; 10 mg/ml) 0.2 μl 11 μl StyI (10 U/μl) 1 μl 55.2 μl Total 14.75 μl 814.2 μl
Add Sty Digestion Master Mix to Samples (stage 1, steps 15-22) - To add the Sty Digestion Master Mix to samples, use a single channel P200 pipet, aliquot 67 μl of Sty Digestion Master Mix to each tube of the strip tubes labeled Dig.
- Using a 12-channel P20 pipet, add 14.75 μl of Sty Digestion Master Mix to each DNA sample in the cooling chamber on ice. The total volume in each well is now 19.75 μl.
Reagent Volume Genomic DNA (50 ng/μl) 5 μl Digestion Master Mix 14.75 μl Total Volume 19.75 μl - Seal the plate tightly with adhesive film.
- Vortex the center of the plate at high speed for 3 sec.
- Spin down the plate at 560 x g for 30 sec.
- Ensure that the lid of thermal cycler is preheated.
- Load the plate onto the thermal cycler and run the GW5.0/6.0 Digest program.
GW5.0/6.0 Digest ProgramTemperature Time 37 °C 120 min 65 °C 20 min 4 °C Hold - When the program is finished, remove the plate and spin it down at 560 x g for 30 sec.
Stage 2: Sty Ligation
During this stage, the digested samples are ligated using the Sty Adaptor.
- Thaw/place the following reagents on ice (approximately 20 min):
- Adaptor StyI
- T4 DNA Ligase Buffer (10x)
- Sty digested samples (if frozen)
- AccuGENE water
- Adaptor StyI
- Label the following tubes, then place in the cooling chamber:
- One strip of 12 tubes labeled “Lig”
- A 2.0 ml Eppendorf tube labeled “Lig MM”
- Solution basin
- One strip of 12 tubes labeled “Lig”
- Prepare the digested samples as follows:
- Vortex the center of the plate at high speed for 3 sec.
- Spin down the plate at 560 x g for 30 sec.
- Place back in the cooling chamber on ice.
- Vortex the center of the plate at high speed for 3 sec.
- To prepare the reagents:
- Vortex at high speed 3 times, 1 sec each time (except for the enzyme).
- Pulse spin for 3 sec.
- Place in the cooling chamber.
- Vortex at high speed 3 times, 1 sec each time (except for the enzyme).
- Preheat the Thermal Cycler lid, however leave the block at room temperature. The lid must be preheated before samples are loaded.
Note: T4 DNA Ligase Buffer (10x) contains ATP and should be thawed on ice. Vortex the buffer as long as necessary before use to ensure precipitate is re-suspended and that the buffer is clear. Avoid multiple freeze-thaw cycles per vendor instructions.
Prepare the Sty Ligation Master Mix (stage 2, steps 6-13) - Keeping all reagents and tubes on ice, prepare the Sty Ligation Master Mix by adding to the 2.0 ml Eppendorf tube the following reagents based on the volumes shown in Table 2:
- Adaptor StyI
- T4 DNA Ligase Buffer (10x)
Table 2. StyI Ligation Master MixReagent 1 Sample 48 Samples (15% extra) T4 Ligase Buffer (10x) 2.5 μl 150 μl Adaptor StyI (50 μM) 0.75 μl 45 μl T4 DNA Ligase (400 U/μl) 2 μl 120 μl Total 5.25 μl 315 μl - Adaptor StyI
- Remove the T4 DNA Ligase from the freezer and immediately place in the cooler on ice.
- Pulse spin the T4 DNA Ligase for 3 sec.
- Immediately add the T4 DNA Ligase to the master mix; then place back in the cooler.
- Vortex the master mix at high speed 3 times, 1 sec each time.
- Pulse spin for 3 sec.
- Place the master mix on ice.
- Proceed immediately to Add Sty Ligation Master Mix to Reactions.
Add Sty Ligation Master Mix to Reactions (stage 2, steps 14-27) - Add Sty Ligation Master Mix to samples using a single channel P100 pipet to aliquot 25 μl of Sty Ligation Master Mix to each tube of the strip tubes on ice.
- Using a 12-channel P20 pipet, aliquot 5.25 μl of Sty Ligation Master Mix to each reaction on the Sty Digestion Stage Plate. The total volume in each well is now 25 μl.
Reagent Volume Sty Digested DNA 19.75 μl Sty Ligation Master Mix 5.25 μl Total 25 μl - Seal the plate tightly with adhesive film.
- Vortex the center of the plate at high speed for 3 sec.
- Spin down the plate at 560 x g for 30 sec.
- Ensure that the thermal cycler lid is preheated.
- Load the plate onto the thermal cycler and run the GW5.0/6.0 Ligate program.
GW5.0/6.0 Ligate ProgramTemperature Time 16 °C 180 min 70 °C 20 min 4 °C Hold - When the GW5.0/6.0 Ligate program is finished, remove the plate and spin it down at 560 x g for 30 sec.
- Place the plate in a cooling chamber on ice.
- Dilute each reaction as follows:
- Pour 10 ml AccuGENE water into the solution basin.
- Using a 12-channel P200 pipet, add 75 μl of the water to each reaction.
Reagent Volume Sty Ligated DNA 25 μl AccuGENE water 75 μl Total 100 μl - Pour 10 ml AccuGENE water into the solution basin.
- Seal the plate tightly with adhesive film.
- Vortex the center of the plate at high speed for 3 sec.
- Spin down the plate at 560 x g for 30 sec.
- Store the plate in a cooling chamber on ice for up to 60 min. If not proceeding directly to the next step, store the plate at -20 °C.
Note: It is crucial to dilute the ligated DNA with AccuGENE water prior to PCR.
Stage 3: Sty PCR
During this stage you will transfer equal amounts of each Sty ligated sample into three fresh 96-well plates; prepare the Sty PCR Master Mix, and add it to each sample; place each plate on a thermal cycler and run the GW6.0 PCR program; and confirm the PCR by running 3 μl of each PCR product on a 2% TBE gel or an E-Gel® 96 2% agarose gel.
- Thaw/place the following reagents on ice (approximately 20 min):
- TITANIUM Taq PCR Buffer
- dNTPs
- PCR Primer 002
- Sty ligated samples (if frozen)
- AccuGENE water
- GC-Melt
- Solution basin
- TITANIUM Taq PCR Buffer
- Label the following tubes, then place in the cooling chamber or on ice, as appropriate:
- Three 96-well reaction plates labeled “P1”, “P2”, “P3”
- One 50 ml Falcon tube labeled “PCR MM”
- Three 96-well reaction plates labeled “P1”, “P2”, “P3”
- Prepare the Sty ligated samples as follows:
- Vortex the center of the plate at high speed for 3 sec.
- Spin down the plate at 560 x g for 30 sec.
- Label the plate Lig.
- Place back in the cooling chamber on ice.
- Vortex the center of the plate at high speed for 3 sec.
- To prepare the reagents:
- Vortex at high speed 3 times, 1 sec each time (except for the enzyme).
- Pulse spin for 3 sec.
- Place in a cooling chamber.
- Vortex at high speed 3 times, 1 sec each time (except for the enzyme).
- Preheat the Thermal Cycler lid, however leave the block at room temperature.
Aliquot Sty Ligated DNA to the PCR Plates (stage 3, steps 6-7) - Working one row at a time and using a 12-channel P20 pipet, transfer 10 μl of each Sty ligated sample to the corresponding well of each PCR plate.
- Seal each plate with adhesive film, and leave in cooling chambers on ice.
Prepare the Sty PCR Master Mix (stage 3, steps 8-13) - To prepare the Sty PCR Master Mix, keep the 50 ml Falcon tube in the cooling chamber and add the reagents as shown in Table 3 (except for the Taq DNA polymerase).
Table 3. Sty PCR Master Mix for 48 SamplesReagent 1 Reaction 3 PCR Pates,
48 Samples each plate
(15% Extra)AccuGENE water 39.5 μl 6.541 ml TITANIUM Taq PCR Buffer (10x) 10 μl 1.656 ml GC-Melt (5 M) 20 μl 3.312 ml dNTP (2.5 mM each) 14 μl 2.318 ml PCR Primer 002 (100 μM) 4.5 μl 0.745 ml TITANIUM Taq DNA Polymerase (50x) (do not add until ready to aliquot master mix to ligated samples) 2 μl 0.331 ml Total 90 μl 14.903 ml - Remove the TITANIUM Taq DNA Polymerase from the freezer and immediately place in a cooler.
- Pulse spin the Taq DNA polymerase for 3 sec.
- Immediately add the Taq DNA polymerase to the master mix; then return the tube to the cooler on ice.
- Vortex the master mix at high speed 3 times, 1 sec each time.
- Pour the mix into the solution basin, keeping the basin on ice.
Add Sty PCR Master Mix to Samples (stage 3, steps 14-18) - To add Sty PCR Master Mix to samples, use a 12-channel P200 pipet and add 90 μl Sty PCR Master Mix to each sample. To avoid contamination, change pipet tips after each dispense. The total volume in each well is 100 μl.
- Seal each reaction plate tightly with adhesive film.
- Vortex the center of each reaction plate at high speed for 3 sec.
- Spin down the plates at 560 x g for 30 sec.
- Keep the reaction plates in cooling chambers on ice until loaded onto the thermal cyclers.
Load Sty PCR Plates onto Thermal Cyclers (stage 3, steps 19-22) - Transfer the plates to the Main Lab.
- Ensure that the thermal cycler lids are preheated. The block should be at room temperature.
- Load each reaction plate onto a thermal cycler.
- Run the GW5.0/6.0 PCR program. The program varies depending upon the thermal cyclers you are using. The example used here is for GeneAmp® PCR System 9700, however another protocol can be found in the manufacturers’ instructions.
GW5.0/6.0 PCR Program for GeneAmp® PCR System 9700Specify Maximum mode.Temperature Time Cycles 94 °C 3 min 1x 94 °C 30 sec | 60 °C 45 sec 30x 68 °C 15 sec | 68 °C 7 min 1x 4 °C Hold Volume: 100 μl
Running Gels (stage 3, steps 23-33)
Note: The instructions below are for running 2% TBE gels. To ensure consistent results, take 3 μl aliquot from each PCR. - When the GW5.0/6.0 PCR program is finished, remove each plate from the thermal cycler.
- Spin down plates at 560 x g for 30 sec.
- Place plates in cooling chambers on ice or keep at 4 °C.
- Label three fresh 96-well reaction plates P1Gel, P2Gel and P3Gel.
- Aliquot 3 μl of 2x Gel Loading Dye to each well in rows A through D of the fresh, labeled PXGel plates.
- Using a 12-channel P20 pipet, transfer 3 μl of each PCR product from the 3 Sty PCR plates to the corresponding plate, row and wells of the PXGel plates. Example: 3 μl of each PCR product from each well of row A on plate P1 is transferred to the corresponding wells of row A on plate P1Gel.
- Seal the PXGel plates.
- Vortex the center of each PXGel plate, then spin them down at 560 x g for 30 sec.
- Load the total volume from each well of each PXGel plate onto 2% TBE gels.
- Run the gels at 120 V for 40 min to 1 h.
- Verify that the PCR product distribution is between ~200 bp to 1,100 bp (example of gel below, Figure 1).
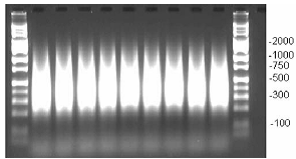
Figure 1. Example of PCR products run on 2% TBE agarose gel at 120 V for 1 h. Average product distribution is between ~200 to 1,100 bp.
Stage 4: Nsp Restriction Enzyme Digestion
During this stage, the genomic DNA is digested by the Nsp I enzyme.
- Thaw/place the following reagents on ice (approximately 20 min):
- NE Buffer 2
- BSA
- Genomic DNA (if frozen)
- AccuGENE water
- NE Buffer 2
- Label the following tubes, then place in the cooling chamber or on ice, as appropriate:
- One strip of 12 tubes labeled “Dig”
- A 2.0 ml Eppendorf tube labeled “Dig MM”
- One strip of 12 tubes labeled “Dig”
- Prepare the plate with genomic DNA as follows:
- Vortex the center of the plate at high speed for 3 sec.
- Spin down the plate at 560 x g for 30 sec.
- Place back in the cooling chamber on ice.
- Vortex the center of the plate at high speed for 3 sec.
- Prepare the reagents (except for the enzyme) as follows:
- Vortex 3 times, 1 sec each time.
- Pulse spin for 3 sec.
- Place in the cooling chamber.
- Vortex 3 times, 1 sec each time.
- Preheat the Thermal Cycler lid, however leave the block at room temperature.
Note: Leave the NSP I enzyme at -20 °C until ready to use.
Prepare the Nsp Digestion Master Mix (stage 4, steps 6-13) - Keeping all reagents and tubes on ice, prepare the Nsp Digestion Master Mix by adding the appropriate volumes of the following reagents from Table 4:
- AccuGENE water
- NE Buffer 2
- BSA
- AccuGENE water
- Remove the Nsp I enzyme from the freezer and immediately place in a cooler.
- Pulse spin the enzyme for 3 sec.
- Immediately add the enzyme to the master mix, then place remaining enzyme back in the cooler.
- Vortex the master mix at high speed 3 times, 1 sec each time.
- Pulse spin for 3 sec.
- Place in the cooling chamber.
- Return any remaining enzyme to the freezer.
Table 4. NspI Digestion Master MixReagent 1 Sample 48 Samples (15% Extra) AccuGENE® Water 11.55 μl 637.6 μl NE Buffer 2 (10x) 2 μl 110.4 μl BSA (100x; 10 mg/ml) 0.2 μl 11 μl NspI (10 U/μl) 1 μl 55.2 μl Total 14.75 μl 814.2 μl
Add Nsp Digestion Master Mix to Samples (stage 4, steps 14-21) - To add Nsp Digestion Master Mix to samples, use a single channel P200 pipet and aliquot 67 μl of Nsp Digestion Master Mix to each tube of the strip tubes labeled Dig.
- Using a 12-channel P20 pipet, add 14.75 μl of Nsp Digestion Master Mix to each DNA sample in the cooling chamber on ice. The total volume in each well is now 19.75 μl.
Reagent Volume Genomic DNA (50 ng/μl) 5 μl Nsp Digestion Master Mix 14.75 μl Total Volume 19.75 μl - Seal the plate tightly with adhesive film.
- Vortex the center of the plate at high speed for 3 sec.
- Spin down the plate at 560 x g for 30 sec.
- Ensure that the lid of thermal cycler is preheated.
- Load the plate onto the thermal cycler and run the GW5.0/6.0 Digest program.
GW5.0/6.0 Digest ProgramTemperature Time 37 °C 120 min 65 °C 20 min 4 °C Hold - When the program is finished, remove the plate and spin it down at 560 x g for 30 sec.
Stage 5: Nsp Ligation
During this stage, the digested samples are ligated using the Nsp Adaptor.
- Thaw/place the following reagents on ice (approximately 20 min):
- Adaptor NspI
- T4 DNA Ligase Buffer (10x)
- Nsp digested samples (if frozen)
- AccuGENE water
- Adaptor NspI
- Label the following tubes, then place in the cooling chamber or on ice, as appropriate:
- One strip of 12 tubes labeled “Lig”
- A 2.0 ml Eppendorf tube labeled “Lig MM”
- Solution basin
- One strip of 12 tubes labeled “Lig”
- Prepare the digested samples as follows:
- Vortex the center of the plate at high speed for 3 sec.
- Spin down the plate at 560 x g for 30 sec.
- Place back in the cooling chamber on ice.
- Vortex the center of the plate at high speed for 3 sec.
- To prepare the reagents:
- Vortex at high speed 3 times, 1 sec each time (except for the enzyme).
- Pulse spin for 3 sec.
- Place in the cooling chamber.
- Vortex at high speed 3 times, 1 sec each time (except for the enzyme).
- Preheat the Thermal Cycler lid, however leave the block at room temperature.
Prepare the Nsp Ligation Master Mix (stage 5, steps 6-12) - Keeping all reagents and tubes on ice, prepare the Nsp Ligation Master Mix by adding to the 2.0 ml Eppendorf tube the following reagents based on the volumes in Table 5.
- Adaptor Nsp
- T4 DNA Ligase Buffer (10x)
- Adaptor Nsp
- Remove the T4 DNA Ligase from the freezer and immediately place in the cooler on ice.
- Pulse spin the T4 DNA Ligase for 3 sec.
- Immediately add the T4 DNA Ligase to the master mix; then place back in the cooler.
- Vortex the master mix at high speed 3 times, 1 sec each time.
- Pulse spin for 3 sec.
- Place the master mix on ice.
Table 5. NspI Ligation Master MixReagent 1 Sample 48 Samples (15% Extra) T4 DNA Ligase Buffer (10x) 2.5 μl 150 μl Adaptor NspI (50 μM) 0.75 μl 45 μl T4 DNA Ligase (400 U/μl) 2 μl 120 μl Total 5.25 μl 315 μl
Add Nsp Ligation Master Mix to Reactions (stage 5, steps 13-25) - To add Nsp Ligation Master Mix to samples, use a single channel P100 pipet and aliquot 25 μl of Nsp Ligation Master Mix to each tube of the strip tubes on ice.
- Using a 12-channel P20 pipet, aliquot 5.25 μl of Nsp Ligation Master Mix to each reaction on the Nsp Digestion Stage Plate. The total volume in each well is now 25 μl.
Reagent Volume Nsp Digested DNA 19.75 μl Nsp Ligation Master Mix 5.25 μl Total 25 μl - Seal the plate tightly with adhesive film.
- Vortex the center of the plate at high speed for 3 sec.
- Spin down the plate at 560 x g for 30 sec.
- Ensure that the thermal cycler lid is preheated.
- Load the plate onto the thermal cycler and run the GW5.0/6.0 Ligate program.
GW5.0/6.0 Ligate ProgramTemperature Time 16 °C 180 min 70 °C 20 min 4 °C Hold - When the GW5.0/6.0 Ligate program is finished, remove the plate and spin it down at 560 x g for 30 sec.
- Place the plate in a cooling chamber on ice.
- Dilute each reaction as follows:
- Pour 10 ml AccuGENE water into the solution basin.
- Using a 12-channel P200 pipet, add 75 μl of the water to each reaction.
The total volume in each well is 100 μl.Reagent Volume Nsp Ligated DNA 25 μl AccuGENE water 75 μl Total 100 μl
- Pour 10 ml AccuGENE water into the solution basin.
- Seal the plate tightly with adhesive film.
- Vortex the center of the plate at high speed for 3 sec.
- Spin down the plate at 560 x g for 30 sec.
Note: It is crucial to dilute the ligated DNA with AccuGENE water prior to PCR.
Stage 6: Nsp PCR
During this stage, you will transfer equal amounts of each Nsp ligated sample into four fresh 96-well plates; prepare the Nsp PCR Master Mix, and add it to each sample; place each plate on a thermal cycler and run the GW6.0 PCR program; and confirm the PCR by running 3 μl of each PCR product on a 2% TBE gel or an E-Gel® 96 2% agarose gel.
- Thaw/place the following reagents on ice (approximately 20 min):
- TITANIUM Taq PCR Buffer
- dNTPs
- PCR Primer 002
- AccuGENE water
- Nsp ligated samples (if frozen)
- GC-Melt
- Solution basin
- TITANIUM Taq PCR Buffer
- Label the following tubes, then place in the cooling chamber or on ice, as appropriate:
- Four 96-well reaction plates labeled “P1”, “P2”, “P3”, “P4”
- One 50 ml Falcon tube labeled “PCR MM”
- Four 96-well reaction plates labeled “P1”, “P2”, “P3”, “P4”
- Place enough cooling chambers for 5 plates and one cooler on ice.
Note: Leave the TITANIUM Taq DNA Polymerase at –20 °C until ready to use. - Prepare the Nsp ligated samples as follows:
- Vortex the center of the plate at high speed for 3 sec.
- Spin down the plate at 560 x g for 30 sec.
- Label the plate Lig.
- Place back in the cooling chamber on ice.
- Vortex the center of the plate at high speed for 3 sec.
- To prepare the reagents:
- Vortex at high speed 3 times, 1 sec each time (except for the enzyme).
- Pulse spin for 3 sec.
- Place in a cooling chamber.
- Vortex at high speed 3 times, 1 sec each time (except for the enzyme).
- Preheat the Thermal Cycler lid, however leave the block at room temperature.
Aliquot Nsp Ligated DNA to the PCR Plates (stage 6, steps 7-8) - To aliquot Nsp ligated DNA to the PCR plates, working one row at a time and using a 12-channel P20 pipet, transfer 10 μl of each Nsp ligated sample to the corresponding well of each PCR plate (P1, P2, P3 and P4).
- Seal each plate with adhesive film, and leave in cooling chambers on ice.
Prepare the Nsp PCR Master Mix (stage 6, steps 9-14) - To prepare the Nsp PCR Master Mix, keep the 50 ml Falcon tube in the cooling chamber and add the reagents as shown in Table 6 (except for the Taq DNA polymerase).
- Remove the TITANIUM Taq DNA Polymerase from the freezer and immediately place in a cooler.
- Pulse spin the Taq DNA polymerase for 3 sec.
- Immediately add the Taq DNA polymerase to the master mix; then return the tube to the cooler on ice.
- Vortex the master mix at high speed 3 times, 1 sec each time.
- Pour the mix into the solution basin, keeping the basin on ice.
Table 6. Prepare the Nsp PCR Master MixReagent For 1 Reaction 4 PCR Plates (15% extra) AccuGENE water 39.5 μl 8.722 ml TITANIUM Taq PCR Buffer (10x) 10 μl 2.208 ml GC-Melt (5M) 20 μl 4.416 ml dNTP (2.5 mM each) 14 μl 3.091 ml PCR Primer 002 (100 μM) 4.5 μl 0.994 ml TITANIUM Taq DNA Polymerase (50x) (do not add until ready to aliquot master mix to ligated samples) 2 μl 0.442 ml Total 90 μl 19.873 ml
Add Nsp PCR Master Mix to Samples (stage 6, steps 15-19) - To add Nsp PCR Master Mix to samples, use a 12-channel P200 pipet and add 90 μl Nsp PCR Master Mix to each sample. To avoid contamination, change pipet tips after each dispense. The total volume in each well is 100 μl.
- Seal each reaction plate tightly with adhesive film.
- Vortex the center of each reaction plate at high speed for 3 sec.
- Spin down the plates at 560 x g for 30 sec.
- Keep the reaction plates in cooling chambers on ice until loaded onto the thermal cyclers.
Load Nsp PCR Plates onto Thermal Cyclers (stage 6, steps 20-22) - To load the plates and run the GW5.0/6.0 PCR program, ensure that the thermal cycler lids are preheated. The block should be at room temperature.
- Load each reaction plate onto a thermal cycler.
- Run the GW5.0/6.0 PCR program. The program varies depending upon the thermal cyclers you are using. The example used here is for GeneAmp® PCR System 9700, however another protocol can be found in the manufacturers’ instructions.
GW5.0/6.0 PCR Program for GeneAmp® PCR System 9700Volume: 100 μlTemperature Time Cycles 94 °C 3 min 1x 94 °C 30 sec | 60 °C 45 sec 30x 68 °C 15 sec | 68 °C 7 min 1x 4 °C Hold
Specify Maximum mode.
Running Gels (stage 6, steps 23-33)
The instructions below are for running 2% TBE gels. To ensure consistent results, take 3 μl aliquot from each PCR.
- When the GW5.0/6.0 PCR program is finished, remove each plate from the thermal cycler.
- Spin down plates at 560 x g for 30 sec.
- Place plates in cooling chambers on ice or keep at 4 °C.
- Label four fresh 96-well reaction plates P1Gel, P2Gel, P3Gel, and P4Gel.
- Aliquot 3 μl of 2x Gel Loading Dye to each well in rows A through D of the fresh, labeled PXGel plates.
- Using a 12-channel P20 pipet, transfer 3 μl of each PCR product from the 4 Nsp PCR plates to the corresponding plate, row and wells of the PXGel plates. Example: 3 μl of each PCR product from each well of row A on plate P1 is transferred to the corresponding wells of row A on plate P1Gel.
- Seal the PXGel plates.
- Vortex the center of each PXGel plate, then spin them down at 560 x g for 30 sec.
- Load the total volume from each well of each PXGel plate onto 2% TBE gels.
- Run the gels at 120 V for 40 min to 1 h.
- Verify that the PCR product distribution is between ~200 bp to 1,100 bp (example of gel below, Figure 2).
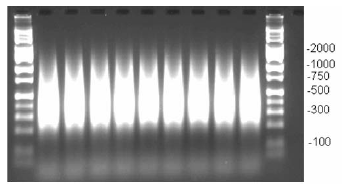
Figure 2. Example of PCR products run on 2% TBE agarose gel at 120 V for 1 h. Average product distribution is between ~200 to 1,100 bp.
Stage 7: PCR Product Purification Using a Millipore Filter Plate
During this stage, you will pool the Sty and Nsp PCR reactions and purify DNA and transfer the purified products to a new 96-well plate.
- Prepare 75% EtOH by diluting ACS-grade or equivalent ethanol to 75% using AccuGENE water.
- Allow the Buffer EB to warm to room temperature prior to use.
- To prepare the manifold, connect the manifold and regulator to a suitable vacuum source able to maintain 20 to 24 in Hg. Leave the vacuum turned off at this time.
- Inspect the manifold for salt and other contaminants and clean if necessary.
- Place the vacuum flask trap below the level of the manifold.
- Place the standard collar on the manifold.
Pool the PCR Products (stage 7, steps 7-14) - If PCR products are frozen, thaw to room temperature on the bench top in plate holders, and if on thermal cyclers, remove them now.
- Vortex the center of each plate at high speed for 3 sec.
- Spin down each plate at 560 x g for 30 sec.
- Place each PCR plate in a plate holder on the bench top.
- Place a deep well pooling plate on the bench top.
- On each PCR plate, cut the seal between each row so that it can be removed one row at a time.
- Using a 12-channel P200 pipet set to 110 μl:
- Remove the seal to expose row A only on each PCR plate.
- Transfer the reactions from row A of each PCR plate to the corresponding wells of row A on the pooling plate.
Sty PCR plates (3): 100 μl from each well = 300 μl/well
Nsp PCR Plate (4): 100 μl from each well = 400 μl/well
Total Volume Each Well of Pooling Plate = 700 μl/well - Change your pipet tips.
- Change pipet tips after the PCR product from the same row of each PCR plate has been pooled on the pooling plate.
- Remove the seal from each PCR plate to expose the next row.
- Transfer each reaction from the same row of each PCR plate to the corresponding row and wells of the pooling plate.
- Repeat steps C., D. and E. until all of the reactions from each PCR plate are pooled.
- Remove the seal to expose row A only on each PCR plate.
- When finished, examine the wells of each PCR plate to ensure that all of the product has been transferred and pooled.
Note: Be very careful when pooling PCR products. Avoid cross-contaminating neighboring wells with small droplets.
Purify the Pooled PCR products (stage 7, steps 15-18) - Mix the magnetic bead stock very well by vigorously shaking the bottle. Beads will settle overnight. Examine the bottom of the bottle and ensure that the solution appears homogenous.
- Pour or pipet 50 ml of magnetic beads to a solution basin. 1 ml of magnetic beads is required for each reaction. You can add in multiple batches if the solution basin is not large enough.
- Using a manual (not electronic) 12-channel P1200 pipet:
- Slowly add 1.0 ml of magnetic beads to each well of pooled PCR product.
- Mix well by pipetting up and down 5 times using the following technique:
- Depress the plunger and place the pipet tips into the top of the solution.
- Move the pipet tips down-aspirating at the same time-until the tips are near the bottom of each well.
- Raise the tips out of the solution.
- Place the pipet tips against the wall of each well just above each reaction, and carefully dispense the solution.
Note: The solution is viscous and sticky. Pipet carefully to ensure that you aspirate and dispense 1 ml. Do not use an electronic pipet. Thorough mixing is critical to ensure that the PCR products bind to the beads. - Change pipet tips for each row.
- Slowly add 1.0 ml of magnetic beads to each well of pooled PCR product.
- Cover the plate to protect the samples from environmental contaminants and allow to incubate at room temperature for 10 min. You can use the lid from a pipet tip box to cover the wells.
Transfer Reactions to a Filter Plate (stage 7, steps 19-21)
- Place the filter plate on the standard collar on the vacuum manifold.
- Using a 12-channel P1200 pipet, transfer each reaction from the pooling plate to the corresponding row and well of the filter plate.
Note: You will need to pipet twice to transfer all of the solution from each well to the filter plate. The solution is viscous and sticky, so check to ensure that all of it has been transferred. - Tightly seal the unused wells with a MicroAmp Clear Adhesive Film. To ensure a tight seal, cover 1/2 to 1/3 of the wells in row D as well. Unused wells must be sealed to ensure proper vacuum pressure.
Purify the Reactions (stage 7, steps 22-30) - To purify the reactions, turn on the vacuum to 20 to 24 in Hg and check the seals. Do not exceed 24 in Hg. Adjust the leak valve if necessary.
- Ensure that the unused wells are completely sealed, and cover the plate to protect it from environmental contaminants.
- Run the vacuum until all of the liquid has been pulled through the filter (approximately 40 to 50 min), then turn off the vacuum.
- Examine each well. There should be no standing liquid in any well, and the wells should appear dull (matte). Wet wells will look shiny. If any of the wells are still wet, put the plate back on the vacuum and continue filtering for up to 10 min (total ≤ 60 min).
- Using a 12-channel P1200 set to 900 μl:
- Add 900 μl of 75% EtOH to each reaction.
- Turn the vacuum on to 20 to 24 in Hg.
- Run the vacuum for approximately 1-2 min (or until the volume in the wells begins to decrease).
- Add another 900 μl of 75% EtOH to each reaction (for a total of 1.8 ml EtOH).
- Cover the plate.
- Run the vacuum until all of the liquid has been pulled through the filter (approximately 10 to 15 min), then turn off the vacuum.
- Add 900 μl of 75% EtOH to each reaction.
- Examine each well. Again, there should be no standing liquid in any well, and the wells should appear dull (matte). Wet wells will look shiny. If any of the wells are still wet, put the plate back on the vacuum and continue filtering for up to 5 min (total ≤ 20 min).
- Remove any excess EtOH as follows:
- Blot the bottom of the plate on Kimwipes.
- Wipe the bottom of the plate with a clean Kimwipe.
- Blot the bottom of the plate on Kimwipes.
- Return the filter plate to the manifold and turn on the vacuum for an additional 10 min only. Do not exceed 10 min. Less than 10 min is ok if no excess ethanol is present in the wells or on the underside of the filter plate.
Note: The purpose of this step is to remove excess EtOH so that it is not carried over into the eluate. Ten minutes is sufficient for this purpose. Leaving the vacuum on for more than 10 min may over-dry the beads which may inhibit elution of the purified DNA. - Turn off the vacuum, and blot the bottom of the plate on Kimwipes to remove any remaining EtOH.
Elute the Purified Reactions (stage 7, steps 31-44) - To elute the purified reactions, attach the elution catch plate to the bottom of the filter plate using 2 strips of lab tape. The filter and elution plate assembly is now referred to as the plate stack.
Note: Do not completely seal with tape. Product will not elute if sealed. - Pour or pipet 3 ml of Buffer EB to a solution basin.
- Using a 12-channel P200 pipet, add 55 μl of Buffer EB to each well. For accurate pipetting, pre-wet pipet tips with EB before dispensing. Dispense as close to the beads as possible without touching them. Buffer EB should be applied directly on top of the beads.
- Tap the plate stack to move all Buffer EB onto the filter at the bottom of the wells.
- Using an adhesive film, tightly seal the filter plate.
- Place the plate stack on a Jitterbug for 10 min at setting 5.
- Inspect each well to verify that the beads are thoroughly resuspended.
- The beads must be thoroughly resuspended in Buffer EB so that the DNA can come off the beads.
- Remove the plate stack from the Jitterbug and remove the adhesive seal.
- Continue elution on the vacuum manifold as follows:
- Remove the standard collar from the manifold.
- Place the plate stack inside the manifold.
- Place the standard collar around the plate stack (Figure 4.13 on page 97).
- Seal the empty wells with adhesive film.
- Turn the vacuum on to 20 to 24 in Hg and ensure that a seal has been formed between the collar and the base of the manifold.
- Ensure that the unused wells are completely sealed.
- Cover the plate stack to protect it from environmental contaminants.
- Run the vacuum until all of the liquid has been pulled through the filter (approximately 5 to 15 min).
- Turn off the vacuum.
- Remove the standard collar from the manifold.
- Examine each well. Again, there should be no standing liquid in any well, and the wells should appear dull (matte). Wet wells will look shiny. If any of the wells are still wet, continue filtering for up to 15 additional min.
- Seal the plate stack with an adhesive film, and spin it down at room temperature for 5 min at 1,400 x g.
- Remove the elution catch plate from the filter plate.
- Using a 12-channel P200 pipet, transfer 45 μl of eluate to a new PCR plate for fragmentation.
Stage 8: Quantitation
During this stage, you will prepare one dilution of each PCR product in optical plates. You will then quantitate the diluted PCR products.
Note: This protocol has been optimized using a UV spectrophotometer plate reader for quantitation. The NanoDrop® will give different quantitation results. This protocol has not been optimized for use with this instrument. In addition, the NanoDrop quantifies a single sample at a time and is not amenable to 96-well plate processing.
- Turn on the spectrophotometer now and allow it to warm for 10 min before use.
- Place the following on the bench top
- Optical plate
- Solution basin
- AccuGENE water
- Optical plate
- Label the optical plate “OP”.
- Prepare the purified, eluted PCR product plate as follows:
- If the plate was frozen, allow it to thaw in a cooling chamber on ice.
- Spin down the plate at 560 x g for 30 sec.
- Place the plate on the bench top.
- If the plate was frozen, allow it to thaw in a cooling chamber on ice.
- To prepare diluted aliquots of the purified samples, pour 15 ml of room temperature AccuGENE water into the solution basin.
Note: One row of wells on the optical plate are used as blanks and contain AccuGENE water only. - Using a 12-channel P200 pipet aliquot 198 μl of water to each well in rows A through E of the optical plate.
- Using a 12-channel P20 pipet:
- Transfer 2 μl of each purified PCR product from rows A through D of the purified sample plate to the corresponding rows and wells of the optical plate. Row E remains water only and will serve as a blank.
- Pipet up and down 2 times after each transfer to ensure that all of the product is dispensed.
- Examine the pipet tips and aliquots before and after each dispense to ensure that exactly 2 μl has been transferred. The result is a 100-fold dilution.
- Transfer 2 μl of each purified PCR product from rows A through D of the purified sample plate to the corresponding rows and wells of the optical plate. Row E remains water only and will serve as a blank.
- Set a 12-channel P200 pipet to 180 μl.
- Mix each sample by pipetting up and down 3 times. Be careful not to scratch the bottom of the plate, or to introduce air bubbles.
- To quantitate the diluted PCR product, measure the OD of each PCR product at 260, 280 and 320 nm. OD280 and OD320 are used as process controls.
- Determine the OD260 measurement for the water blank and average.
- Determine the concentration of each PCR product as follows:
- Take 1 OD reading for every sample.
OD = (sample OD)-(average water blank OD) - Calculate the undiluted sample concentration for each sample using the Sample OD:
Sample concentration in μg/μl = OD x 0.05 μg/μl x 100
Apply the convention that 1 absorbance unit at 260 nm equals 50 μg/ml (equivalent to 0.05 μg/μl) for double-stranded PCR products. This convention assumes a path length of 1 cm. Consult your spectrophotometer handbook for further information.
- Take 1 OD reading for every sample.
Stage 9: Fragmentation
During this stage, the purified PCR products are fragmented using Fragmentation Reagent. You will first dilute the Fragmentation Reagent by adding the appropriate amount of Fragmentation Buffer and molecular biology-grade water. You will then quickly add the diluted reagent to each reaction, place the plate onto a thermal cycler, and run the GW6.0 Fragment program. Once the program is finished, you will verify fragmentation by running 1.5 μl of each reaction on a 4% TBE gel or an E-Gel 48 4% agarose gel.
- Thaw/place the following reagents on ice (approximately 20 min):
- Fragmentation Buffer
- AccuGENE water
- Fragmentation Buffer
- Prepare the Fragmentation Buffer as follows:
- Vortex 3 times, 1 sec each time.
- Pulse spin for 3 sec.
- Place the buffer in the cooling chamber on ice.
- Vortex 3 times, 1 sec each time.
- Label the following tubes, then place in the cooling chamber or on ice, as appropriate:
- Two strips of 12 tubes each: one labeled ‘Buffer’ and one labeled ‘FR’
- One 2.0 ml Eppendorf tube labeled ‘Frag MM’
- Plate of purified PCR product from the previous stage
- Two strips of 12 tubes each: one labeled ‘Buffer’ and one labeled ‘FR’
- Power on the thermal cycler and preheat the block to 37 °C. Allow it to heat for 10 min before loading samples.
- To prepare the samples for Fragmentation, aliquot 28 μl of 10x Fragmentation Buffer to each tube of the strip tubes labeled Buffer.
- Using a 12-channel P20 pipet, add 5 μl of Fragmentation Buffer to each sample in the 96-well reaction plate. Check your pipet tips each time to ensure that all of the buffer has been dispensed. The total volume in each well is now 50 μl.
- Read the Fragmentation Reagent tube label and record the concentration.
- Dilute the Fragmentation Reagent to 0.1 U/μl as described below using the appropriate recipe from Table 7.
Table 7. Dilution Recipes for the Fragmentation ReagentReagent Fragmentation Reagent Concentration 2 U/μl 2.25 U/μl 2.5 U/μl 2.75 U/μl 3 U/μl AccuGENE water 306 μl 308 μl 309.6 μl 310.9 μl 312 μl 10x Fragmentation Buffer 36 μl 36 μl 36 μl 36 μl 36 μl Fragmentation Reagent 18 μl 16 μl 14.4 μl 13.1 μl 12 μl Total
(enough for 48 samples)360 μl 360 μl 360 μl 360 μl 360 μl - To the 2.0 ml Eppendorf tube on ice, add the AccuGENE water and Fragmentation Buffer. Allow to cool on ice for 5 min.
- Remove the Fragmentation Reagent from the freezer and immediately pulse spin for 3 sec then place in a cooler.
- Add the Fragmentation Reagent to the 1.5 ml Eppendorf tube.
- Vortex the diluted Fragmentation Reagent at high speed 3 times, 1 sec each time.
- Pulse spin for 3 sec and immediately place on ice.
- To add diluted Fragmentation Reagent to the samples, quickly and on ice, aliquot 28 μl of diluted Fragmentation Reagent to each tube of the strip tubes labeled FR. Avoid introducing air bubbles at the bottom of the strip tubes to ensure the accurate transfer of 5 μl diluted DNA to each sample.
- Using a 12-channel P20 pipet, add 5 μl of diluted Fragmentation Reagent to each sample. Do not pipet up and down.
Sample with Fragmentation Buffer 50 μl Diluted Fragmentation Reagent (0.1 U/μl) 5 μl Total 55 μl - Seal the plate and inspect the edges to ensure that it is tightly sealed.
- Vortex the center of the plate at high speed for 3 sec.
- Place the plate in a chilled plastic plate holder and spin it down at 4 °C at 560 x g for 30 sec.
- Immediately load the plate onto the pre-heated block of the thermal cycler (37 °C) and run the GW5.0/6.0 Fragment program.
GW5.0/6.0 Fragment ProgramTemperature Time 37 °C 35 min 95 °C 15 min 4 °C Hold - Discard any remaining diluted Fragmentation Reagent. Diluted Fragmentation Reagent should never be reused.
- Remove the plate from the thermal cycler.
- Spin down the plate at 560 x g for 30 sec, and place in a cooling chamber on ice.
- Dilute 1.5 μl of each fragmented PCR product with 4 μl gel loading dye.
- Run on 4% TBE gel with the BioNexus All Purpose Hi-Lo ladder at 120 V for 30 min to 1 h.
- Inspect the gel (example of gel below, Figure 3).
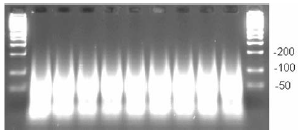
Figure 3. Typical example of fragmented PCR products run on 4% TBE agarose gel at 120 V for 30 min to 1 h. Average fragment size is < 180 bp.
Stage 10: Labeling
During this stage, 48 fragmented samples are labeled using the DNA Labeling Reagent.
- Prepare Your Work Area
To prepare the work area:- Place a double cooling chamber and a cooler on ice.
- Prepare the reagents as follows:
- Vortex each reagent at high speed 3 times, 1 sec each time.
- Pulse spin for 3 sec; then place in the cooling chamber.
Reagents required for Stage 10: LabelingQuantity Reagent 1 vial DNA Labeling Reagent (30 mM) 1 vial Terminal Deoxynucleotidyl Transferase (TdT; 30 U/μl) 1 vial Terminal Deoxynucleotidyl Transferase Buffer (TdT Buffer; 5x)
- Vortex each reagent at high speed 3 times, 1 sec each time.
- Place a double cooling chamber and a cooler on ice.
- Thaw/place the following reagents on ice (approximately 20 min):
- 5x TdT Buffer
- DNA Labeling Reagent
- 5x TdT Buffer
- Label one 15 ml centrifuge tube MM, and place on ice.
- Label and place the following in the cooling chamber:
- One strip of 12 tubes labeled MM
- Plate of fragmented reactions from the previous stage
- One strip of 12 tubes labeled MM
- Power on the thermal cycler and preheat the block to 37 °C. Allow it to heat for 10 min before loading samples.
- To prepare the Labeling Master Mix, add the following to the 15 ml centrifuge tube on ice using the volumes shown in Table 8.
- 5x TdT Buffer
- DNA Labeling Reagent
Reagent 1 Sample 48 Samples (15% extra) TdT Buffer (5x) 14 μl 772.8 μl DNA Labeling Reagent (30 mM) 2 μl 110.4 μl TdT enzyme (30 U/μl) 3.5 μl 193.2 μl Total 19.5 μl 1076.4 μl - 5x TdT Buffer
- Remove the TdT enzyme from the freezer and immediately place in the cooler.
- Pulse spin the enzyme for 3 sec; then immediately place back in the cooler.
- Add the TdT enzyme to the master mix.
- Vortex the master mix at high speed 3 times, 1 sec each time.
- Pulse spin for 3 sec.
- To add the Labeling Master Mix to the samples, and while keeping samples in the cooling chamber and all tubes on ice when making additions, aliquot 89 μl of Labeling Master Mix to each tube of the strip tubes.
- Add the Labeling Master Mix as follows:
- Using a 12-channel P20 pipet, aliquot 19.5 μl of Labeling Master Mix to each sample.
- Pipet up and down one time to ensure that all of the mix is added to the samples. The total volume in each well is now 73 μl.
Reagent Volume Fragmented DNA (less 1.5 μl for gel analysis) 53.5 μl Labeling Mix 19.5 μl Total 73 μl - Using a 12-channel P20 pipet, aliquot 19.5 μl of Labeling Master Mix to each sample.
- Seal the plate tightly with adhesive film.
- Vortex the center of the plate at high speed for 3 sec.
- Spin down the plate at 560 x g for 30 sec.
- Place the plate on the pre-heated thermal cycler block, and run the GW5.0/6.0 Label program.
GW5.0/6.0 Label ProgramTemperature Time 37 °C 4 h 95 °C 15 min 4 °C Hold - When the GW5.0/6.0 Label program is finished:
- Remove the plate from the thermal cycler.
- Spin down the plate at 560 x g for 30 sec.
- Remove the plate from the thermal cycler.
Stage 11: Target Hybridization
During this stage, each reaction is loaded onto a Genome-Wide Human SNP Array 6.0.
- To prepare 1,000 ml of 12x MES Stock Solution: (1.25 M MES, 0.89 M [Na+]), combine:
- 70.4 g MES hydrate
- 193.3 g MES sodium salt
- 800 ml AccuGENE® water
- 70.4 g MES hydrate
- Mix and adjust volume to 950 ml.
- Test the pH, which should be between 6.5 and 6.7.
- Adjust the pH so it falls between 6.5 and 6.7.
- Adjust the volume to 1,000 ml.
- Filter the solution through a 0.2 μm filter.
- Protect from light using aluminum foil and store at 4 °C.
- Preheat the hybridization ovens by turning each oven on and setting the temperature to 50 °C.
- Set the rpm to 60.
- Turn the rotation on and allow to preheat for 1 h before loading arrays.
- If the labeled samples from the previous stage were frozen, thaw the plate on the bench top.
- Vortex the center of the plate at high speed for 3 sec.
- Spin down the plate at 560 x g for 30 sec.
- Place in a cooling chamber on ice.
- Labeled samples must be placed in a Bio-Rad unskirted 96-well plate.
- Power on the thermal cycler to preheat the lid. Leave the block at room temperature.
- To prepare the arrays, unwrap the arrays and place on the bench top, septa-side up.
- Mark each array with a meaningful designation (e.g., a number) to ensure that you know which sample is loaded onto each array.
- Allow the arrays to warm to room temperature by leaving on the bench top 10 to 15 min.
- Insert a 200 μl pipet tip into the upper right septum of each array.
- To prepare the Hybridization Master Mix to the 50 ml centrifuge tube, add the reagents in the order shown in Table 9 and mix well.
Note: DMSO addition: pipet directly into the solution of other reagents. Avoid pipetting along the side of the tube.
Table 9. Hybridization Master MixReagent 1 Array 48 Arrays (15% extra) MES (12x; 1.25 M) 12 μl 660 μl Denhardt’s Solution (50x) 13 μl 715 μl EDTA (0.5 M) 3 μl 165 μl HSDNA (10 mg/ml) 3 μl 165 μl OCR, 0100 2 μl 110 μl Human Cot-1 DNA® (1 mg/ml) 3 μl 165 μl Tween-20 (3%) 1 μl 55 μl DMSO (100%) 13 μl 715 μl TMACL (5 M) 140 μl 7.7 ml Total 190 μl 10.45 ml
Using a GeneAmp® PCR System 9700 - To add Hybridization Master Mix and denature the samples, pour 11 ml Hybridization Master Mix into a solution basin.
- Using a 12-channel P200 pipet, add 190 μl of Hybridization Master Mix to each sample on the Label Plate. Total volume in each well is 263 μl.
- Seal the plate tightly with adhesive film.
- Vortex the center of the plate for 30 sec.
- Spin down the plate at 560 x g for 30 sec.
- Cut the adhesive film between each row of samples. Do not remove the film.
- Place the plate onto the thermal cycler and close the lid.
- Run the GW5.0/6.0 Hyb program.
GW5.0/6.0 Hyb ProgramNote: The following procedure requires 2 operators working simultaneously. Operator 1 loads the samples onto the arrays; Operator 2 covers the septa with Tough-Spots and loads the arrays into the hybridization ovens.Temperature Time 95 °C 10 min 49 °C Hold
30a.Operator 1 Tasks (after completing stage 11, step 29)- When the plate reaches 49 °C, slide back the lid on the thermal cycler enough to expose the first row of samples only.
- Remove the film from the first row.
- Using a single-channel P200 pipet, remove 200 μl of denatured sample from the first well.
- Immediately inject the sample into an array.
- Pass the array to Operator 2.
- Remove 200 μl of sample from the next well and immediately inject it into an array.
- Pass the array to Operator 2.
- Repeat this process one sample at a time until the entire row is loaded.
- Place a fresh strip of adhesive film over the completed row.
- Slide the thermal cycler lid back to expose the next row of samples.
- Repeat steps 3 through 10 until all of the samples have been loaded onto arrays.
- Cover the septa on each array with a Tough-Spot.
- For every 4 arrays:
- Load the arrays into an oven tray evenly spaced.
- Immediately place the tray into the hybridization oven.
- Load the arrays into an oven tray evenly spaced.
Notes for Operators 1 and 2:- Load no more than 32 arrays in one hybridization oven at a time.
- All 48 samples should be loaded within 1 h.
- Store the remaining samples and any samples not yet hybridized in a tightly sealed plate at -20 °C.
- Allow the arrays to rotate at 50 °C, 60 rpm for 16 to 18 h.
- Allow the arrays to rotate in the hybridization ovens for 16 to 18 h at 50 °C and 60 rpm. This temperature is optimized for this product, and should be stringently followed.
- Continue on to washing, staining and scanning arrays.
- When the plate reaches 49 °C, slide back the lid on the thermal cycler enough to expose the first row of samples only.
Recipes
- Clontech TITANIUMTM DNA Amplification Kit
- dNTPs (2.5 mM each)
- GC-Melt (5 M)
- TITANIUMTM Taq DNA Polymerase (50x)
- TITANIUMTM Taq PCR Buffer (10x)
- dNTPs (2.5 mM each)
- 12x MES Stock Solution [1.25 M MES, 0.89 M (Na+)]
- 70.4 g MES hydrate
- 193.3 g MES sodium salt
- 800 ml AccuGENE® water
- 70.4 g MES hydrate
Acknowledgments
Funding: Swiss National Foundation 144082 and Gertrude von Meissner Foundation grant.
References
- Komura, D., Shen, F., Ishikawa, S., Fitch, K. R., Chen, W., Zhang, J., Liu, G., Ihara, S., Nakamura, H., Hurles, M. E., Lee, C., Scherer, S. W., Jones, K. W., Shapero, M. H., Huang, J. and Aburatani, H. (2006). Genome-wide detection of human copy number variations using high-density DNA oligonucleotide arrays. Genome Res 16(12): 1575-1584.
Article Information
Copyright
© 2016 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Bosman, A. (2016). Affymetrix Genome-wide Human SNP Assay. Bio-protocol 6(10): e1806. DOI: 10.21769/BioProtoc.1806.
Category
Molecular Biology > DNA > Microarray
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.