- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

ACE-score-based Analysis of Temporal miRNA Targetomes During Human Cytomegalovirus Infection Using AGO-CLIP-seq

Published: Vol 6, Iss 8, Apr 20, 2016 DOI: 10.21769/BioProtoc.1791 Views: 13205
Reviewed by: Yannick DebingFeng LiVaibhav B Shah
Original research article
The authors used this protocol in:

Jun 2015
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics









Abstract
Although temporal regulation of gene expression during the course of infection is known to be critical for determining the outcome of host-virus interactions, systematic temporal analysis of the miRNA targetomes during productive viral infection has been technically challenging due to the large range of miRNA-mRNA cross-talks at the host-virus interface. High-confidence quantifying models of the suppression efficacy in targeting sites by integrating bioinformatics with Argonaute-crosslinking and immunoprecipitation followed by high-throughput sequencing (AGO-CLIP-seq) (Chi et al., 2009) data have been poorly developed. To accurately identify miRNA target sites and calculate the targeting efficacy of miRNA-target interactions, we developed a new bioinformatic quantitation method, AGO-CLIP-seq enrichment (ACE)-scoring algorithm (Kim et al., 2015). Inclusion of the uninfected control in our AGO-CLIP-seq analysis can significantly improve the accuracy of authentic target site identification for viral or human miRNAs and extract physiologically significant changes during productive human cytomegalovirus (HCMV) infection using our ACE-scoring method. Thus, we suggest that our new ACE-scoring-based methodology can be applied to various miRNA targetome studies, which will be performed in other kinds of temporal contexts, such as developmental stages, immune stimulation by cytokines or pathogens, and lytic infection by other viruses.
Keywords: CLIPseqMaterials and Reagents
- 100 mm cell culture dish (Sarstedt AG & Co, catalog number: 83.3902 )
- Disposable serological pipettes (Orange)
- Filter micropipette tips (Biotix, NEPTUNE)
- 1.6 ml microcentrifuge tube (Biotix, NEPTUNE, catalog number: 3745X )
- Cell scraper (25 cm) (Sarstedt AG & Co, catalog number: 83.1830 )
- Plastic wrap (CLEANWRAP)
- BAS film (Fujifilm Corporation)
- Razor (Dorco, model: Pace Single Edge Blades DN52 )
- Costar Spin-X centrifugation column (Sigma-Aldrich, catalog number: CLS8162-96EA )
- Human foreskin fibroblast (HFF) cells (ATCC, catalog number: SCRC-104 )
- Dulbecco’s modified Eagle’s medium (DMEM) (with High Glucose, with 4 mM L-Glutamine, without Sodium Pyruvate) (GE Healthcare, HyCloneTM, catalog number: SH30022.FS )
- Fetal bovine serum (FBS) (Enhance growth of cells with U.S. sourced GE Healthcare HyClone characterized) (GE Healthcare, HyCloneTM, catalog number: SH30071.03HI )
- GlutaMAX-I (100x) (100 ml) (Thermo Fisher Scientific, GibcoTM, catalog number: 35050-061 )
- Penicillin/Streptomycin solution (P/S) (100x, 10,000 U/ml) (Thermo Fisher Scientific, GibcoTM, catalog number: 15-140-122 )
- Ice
- Towne strain of human cytomegalovirus (HCMV) (ATCC, model: VR-977 ; NCBI accession number: FJ616285.1)
- Dynabeads® Sheep-Anti Mouse IgG (Thermo Fisher Scientific, InvitrogenTM, catalog number: 11031 )
- Dynabeads® Pan Mouse IgG (Thermo Fisher Scientific, InvitrogenTM, catalog number: 11041 )
- Pan anti-AGO mAb 2A8 (Diagenode, catalog number: C15200167-100 )
- Protease inhibitor cocktail (Cell Signaling Technology, catalog number: 5871 )
- Recombinant DNase I (Takara Bio Company, catalog number: 2270B )
- RNase A (1 MG, 20 units/μl; 5 mg/ml) (Thermo Fisher Scientific, Affymetrics, catalog number: 70194Y )
- Alkaline Phosphatase, Calf Intestinal (CIP) (New England Biolabs, catalog number: M0290S )
- 3’-Adaptor (RA3) (5’-rApp-TGGAATTCTCGGGTGCCAAGG-3’-ddC, 5’-Adenylation, 3’-Dideoxy-C) (Integrated DNA Technologies, RNase Free HPLC Purification)
- T4 RNA Ligase 2, truncated K227Q (New England Biolabs, catalog number: M0351L )
- SUPERase.In RNase Inhibitor (Thermo Fisher Scientific, AmbionTM, catalog number: AM2696 )
- ATP, [γ-32P], 6,000 Ci/mmol (222 TBq/mmol) (PerkinElmer, catalog number: NEG502Z500UC )
- T4 Polynucleotide Kinase (PNK) (Takara Bio Company, catalog number: 2021A )
- 10 mM Adenosine 5’-Triphosphate (ATP) (New England Biolabs, catalog number: P0756S )
- NuPAGE® LDS Sample Buffer (4x) (Thermo Fisher Scientific, NovexTM, catalog number: NP0007 )
- NuPAGE® Sample Reducing Agent (10x) (Thermo Fisher Scientific, NovexTM, catalog number: NP0004 )
- NuPAGE® Antioxidant (Thermo Fisher Scientific, NovexTM, catalog number: NP0005 )
- NuPAGETM NovexTM 4-12% Bis-Tris Protein Gels, 1.0 mm, 10-well (Thermo Fisher Scientific, InvitrogenTM, catalog number: NP0321BOX )
- NuPAGE® MOPS SDS Running Buffer (20x) (Thermo Fisher Scientific, NovexTM, catalog number: NP0001 )
- NuPAGE® Transfer Buffer (20x) (Thermo Fisher Scientific, NovexTM, catalog number: NP0006 )
- Amersham Protran Supported 0.45 NC (GE Healthcare Life Sciences, catalog number: 10600016 )
- Bovine serum albumin (BSA) (life science grade powder) (Merck Millipore Corporation, Probumin®, catalog number: 821006 )
- Anti-AGO mAb 21D2 (Made in laboratory of Narry V. Kim, School of Biological Sciences, Seoul National University, Seoul, Republic of Korea)
- Peroxidase AffiniPure Goat Anti-Mouse IgG (H+L) (Jackson ImmunoResearch Inc., catalog number: 115-035-003 )
- EMD Millipore ImmobilonTM Western Chemiluminescent HRP Substrate (ECL) (Thermo Fisher Scientific, catalog number: WBKLS0050 )
- Acid-Phenol:Chloroform, pH 4.5 (with IAA, 125:24:1) (RNA phenol/chloroform) (Thermo Fisher Scientific, AmbionTM, catalog number: AM9720 )
- Linear acrylamide (Thermo Fisher Scientific, InvitrogenTM, catalog number: AM9520 )
- 3 M Sodium acetate (pH 5.5) (Thermo Fisher Scientific, AmbionTM, catalog number: AM9740 )
- Ethanol (Merck Millipore Corporation, catalog number: 100983 )
- Isopropanol (2-propanol) (Merck Millipore Corporation, catalog number: 109634 )
- UltraPureTM DNase/RNase-Free Distilled Water (D.W.) (Thermo Fisher Scientific, InvitrogenTM, catalog number: 10977-015 )
- 2x RNA loading buffer (Thermo Fisher Scientific, AmbionTM, catalog number: AM8546G )
- mirVanaTM Probe & Marker Kit (Thermo Fisher Scientific, AmbionTM, catalog number: AM1554 )
- 10% Novex® TBE-Urea Gels, 10 well (Thermo Fisher Scientific, InvitrogenTM, catalog number: EC6875BOX )
- T4 RNA ligase (Takara Bio Company, catalog number: 2050B )
- Proteinase K (PK) (Thermo Fisher Scientific, Macherey-Nagel, catalog number: 740506 )
- 5’-adaptor oligonucleotide (5’-Solexa adaptor)
5’-rGrUrUrCrArGrArGrUrUrCrUrArCrArGrUrCrCrGrArCrGrArUrC-3’ (Integrated DNA Technologies, RNase Free HPLC Purification) - RNA RT primer (RTP)
5’-GCCTTGGCACCCGAGAATTCCA-3’ (Integrated DNA Technologies, RNase Free HPLC Purification) - 5’-Primer (Integrated DNA Technologies, RNase Free HPLC Purification)
- RNA PCR Primer (RP1)
5’-AATGATACGGCGACCACCGAGATCTACACGTTCAGAGTTCTACAGTCCGA-3’
- RNA PCR Primer (RP1)
- 3’-Primers (Integrated DNA Technologies, RNase Free HPLC Purification)
- RNA PCR Primer, Index 1 (RPI1)
5’-CAAGCAGAAGACGGCATACGAGATCGTGATGTGACTGGAGTTCCTTGGCACCCGAGAATTCCA-3’ - RNA PCR Primer, Index 2 (RPI2)
5’-CAAGCAGAAGACGGCATACGAGATACATCGGTGACTGGAGTTCCTTGGCACCCGAGAATTCCA-3’ - RNA PCR Primer, Index 3 (RPI3)
5’-CAAGCAGAAGACGGCATACGAGATGCCTAAGTGACTGGAGTTCCTTGGCACCCGAGAATTCCA-3’ - RNA PCR Primer, Index 4 (RPI4)
5’-CAAGCAGAAGACGGCATACGAGATTGGTCAGTGACTGGAGTTCCTTGGCACCCGAGAATTCCA-3’
- RNA PCR Primer, Index 1 (RPI1)
- Phusion® High-Fidelity DNA Polymerase (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: F-530L )
- dNTP mixture (Enzynomics, catalog number: N001L )
- SuperScriptTM III reverse transcriptase (RT) (Thermo Fisher Scientific, InvitrogenTM, catalog number: 18080-044 )
- Novex® TBE Gels, 6%, 10 Well (Thermo Fisher Scientific, InvitrogenTM, catalog number: EC6265BOX )
- Low Molecular Weight DNA Ladder (New England Biolabs, catalog number: N3233L )
- Ethidium bromide (EtBr) (Sigma-Aldrich, catalog number: E7637 )
- Dynabeads® mRNA DIRECTTM Kit (Thermo Fisher Scientific, AmbionTM, catalog number: 61011 )
- NEBNext® Magnesium RNA Fragmentation Module (New England Biolabs, catalog number: E6150S )
- Sodium chloride (NaCl) (BioXtra, ≥99.5%) (Sigma-Aldrich, catalog number: S7653 )
- Potassium chloride (KCl) (BioXtra, ≥99.0%) (Sigma-Aldrich, catalog number: P9333 )
- Sodium phosphate dibasic (Na2HPO4) (BioXtra, ≥99%) (Sigma-Aldrich, catalog number: S7907 )
- Potassium phosphate monobasic (KH2PO4) (≥99.0%) (Sigma-Aldrich, catalog number: P5655 )
- Trizma® base (An high-quality Tris base) (BioXtra, ≥99.9%) (Sigma-Aldrich, catalog number: T6791 )
- NP-40 Alternative (Merck Millipore Corporation, catalog number: 492018 )
- Sodium dodecyl sulfate (SDS) [BioXtra, ≥99.0% (GC)] (Sigma-Aldrich, catalog number: L6026 )
- Sodium deoxycholate (BioXtra, anhydrous, ≥98%) (Sigma-Aldrich, catalog number: 30970 )
- Ethylenediaminetetraacetic acid (EDTA) (BioUltra, anhydrous, ≥98%) (Sigma-Aldrich, catalog number: EDS )
- Magnesium chloride (MgCl2) (anhydrous, ≥98%) (Sigma-Aldrich, catalog number: M8266 )
- Tween-20 (Sigma-Aldrich, catalog number: P7949 )
- NEBuffer3 (New England Biolabs, catalog number: B7003S )
- 10x T4 RNA ligase buffer (New England Biolabs, catalog number: B0216L )
- 50% PEG8000 (supplied with 10x T4 RNA ligase buffer)
- 10 mM Adenosine 5’-Triphosphate (ATP) (supplied with 10x T4 RNA ligase buffer)
- Bromophenol blue (ACS reagent) (Sigma-Aldrich, catalog number: 114391 )
- Xylene cyanol FF (BioReagent) (Sigma-Aldrich, catalog number: X4126 )
- Glycerol (BioXtra, anhydrous, ≥99%) (Sigma-Aldrich, catalog number: G6279 )
- Boric acid (BioReagen, ≥99.5%) (Sigma-Aldrich, catalog number: B6768 )
- Trizol® reagent (Thermo Fisher Scientific, AmbionTM, catalog number: 15596018 )
- Dynabeads® mRNA DIRECTTM Purification Kit (Thermo Fisher Scientific, AmbionTM, catalog number: 61011)
- NEBNext® Magnesium RNA Fragmentation Module (New England Biolabs, catalog number: E6150S)
- Antarctic phosphatase (New England Biolabs, catalog number: M0289S )
- Phosphate-buffered saline (PBS) (see Recipes)
- Lysis buffer (see Recipes)
- High-salt buffer (see Recipes)
- Wash buffer (see Recipes)
- CIP mix (see Recipes)
- 3’-ligation mix (see Recipes)
- Hot PNK mix (see Recipes)
- 0.1% PBS-T (see Recipes)
- PK buffer (see Recipes)
- PK mix (see Recipes)
- PK/Urea buffer (see Recipes)
- 5’-ligation mix (see Recipes)
- PCR mix (see Recipes) (see Recipes)
- 6x DNA Loading buffer (see Recipes)
- TBE (Tris-borate-EDTA) (see Recipes)
Equipment
- CO2 incubator (LabX, Sanyo, model: CO2 Incubator MCO-18AIC )
- Micropipettes (Gilson, PIPETMAN ClassicTM P10, P20, P200 and P1000, catalog number: F144802 , F123600 , F123601 and F123602 )
- Rocker (FINEPCR, model: CR300 )
- UV crosslinker (Spectronics Corporation, model: XL1500 )
- Vortexer (Scientific Industries, model: Vortex-Genie® 2 and catalog number: SI-0256 )
- DynaMagTM-2 Magnet (Thermo Fisher Scientific, catalog number: 12321D )
- Rotator (FINEPCR, model: AG) with an accessary Roller Drum (FINEPCR, model: RD25-42 )
- Eppendorf® Thermomixer® R, dry block heating and cooling shaker (Sigma-Aldrich, catalog number: T3317 )
- Refrigerated tabletop microcentrifuge (Hanil BioMed Inc., model: Centrifuge Micro 17TR )
- XCell SureLock® Mini-Cell (Thermo Fisher Scientific, NovexTM, catalog number: EI0001 )
- Chemidoc (Vilber Lourmat, model: Fusion Spectra )
- BAS 2500 (Fujifilm Corporation)
- Deep freezer (-80 °C freezer)
- PCR cycler (MyCyclerTM Thermal Cycler System) (Bio-Rad Laboratories, catalog number: 1709703 )
- Illumina HiSeq2000
Software
- CutAdap software
- Human genome (hg19) BLAST+ (v. 2.2.27) software (Camacho et al., 2009)
Procedure

Figure 1. Scheme of the entire procedure (A) and AGO-CLIP-seq (B)
Note: Throughout the entire AGO-CLIP-seq protocol (Figure 1), all work must be performed in RNase-free environments and only sterile and DNase-/RNase-free solutions and materials should be used.
- AGO-CLIP-seq
- Cell culture and HCMV infection
Note: To propagate HCMV and prepare the HCMV stock, we have followed the previous instruction (Britt, 2010).- HFF cells are obtained from the American Type Culture Collection and cultured in five 100-mm culture dishes with complete DMEM supplemented with 10% FBS, 2 mM GlutaMAX-I, and 1x P/S at 37 °C in the presence of 5% CO2.
- Wash twice with PBS.
- Add HCMV-containing serum-free DMEM at an MOI of 3 with a total volume of 2 ml/dish and incubate at 37 °C for 1 h rocking by hand gently but thoroughly at an interval of 15 min.
- Decant the virus media, wash twice with PBS and add 10 ml 10% FBS, P/S, L-Glu DMEM/dish.
- Incubate for the indicated time (24, 48 and 72 h).
- HFF cells are obtained from the American Type Culture Collection and cultured in five 100-mm culture dishes with complete DMEM supplemented with 10% FBS, 2 mM GlutaMAX-I, and 1x P/S at 37 °C in the presence of 5% CO2.
- UV irradiation
- Wash once with 3 ml/dish of Cold PBS.
- Add 2 ml/dish of Cold PBS in four of five dishes and extract total RNAs from one of five dish to construct libraries for mRNA-seq and smallRNA-seq (go to step B1a).
- Irradiate with 254-nm UV at 250 mJ/cm2 on ice in Spectrolinker.
- Decant PBS completely.
- Prepared cells were stored at -80 °C until subsequent steps.
- Wash once with 3 ml/dish of Cold PBS.
- Preparation of antibody-conjugated Dynabeads
- Transfer 50 µl of Sheep α-mouse immunoglobulin G (IgG) conjugated Dynabead per sample into 1.5 ml tube. To collect the magnetic Dynabeads the DynaMeg-2 magnet rack is used. Pan α-mouse IgG conjugated Dynabead can be used as an alternative for Sheep α-mouse immunoglobulin G (IgG) conjugated Dynabead.
- Wash twice with 0.1 M NaHPO4 (pH 8.1) and resuspend beads in 200 µl 0.1 M NaHPO4 (pH 8.1).
- Add 30 μg of mAb 2A8 as a pan-anti-AGO antibody per sample.
- Rotate the tubes at room temperature for 2 h (at least 1 h).
- Wash twice with Lysis buffer.
- Leave in ice until the next step.
- Transfer 50 µl of Sheep α-mouse immunoglobulin G (IgG) conjugated Dynabead per sample into 1.5 ml tube. To collect the magnetic Dynabeads the DynaMeg-2 magnet rack is used. Pan α-mouse IgG conjugated Dynabead can be used as an alternative for Sheep α-mouse immunoglobulin G (IgG) conjugated Dynabead.
- Cell lysis and partial RNA digestion
- Add 1 ml Lysis buffer (with Protease inhibitor cocktail) into the sample dish, scrap the cell lysate, transfer the lysate into the next dish and repeat until the last dish.
- Finally, transfer the lysate into a 1.5 ml tube.
- Add 10 µl recombinant DNase I and incubate at 37 °C and 1,400 rpm in Eppendorf® Thermomixer® R for 10 min to make the lysate less viscous.
- Add 10 µl of diluted (1:1,000) RNase A.
- Incubate at 37 °C and 1,400 rpm for 10 min.
- Immediately transfer the tubes into ice.
- Centrifuge at 4 °C and 28,000 x g for 30 min.
- Carefully collect the supernatant and transfer the supernatant, except for 80 µl which will be used for immunoblotting, into a new tube to be combined with the prepared beads in the next step.
- Add 1 ml Lysis buffer (with Protease inhibitor cocktail) into the sample dish, scrap the cell lysate, transfer the lysate into the next dish and repeat until the last dish.
- Immunoprecipitation
- Combine prepared Ab-conjugated beads with cell lysates.
- Rotate at 4 °C for 3 h.
- Decant the supernatant and wash the beads twice with 1 ml High-salt buffer.
- Wash twice with 1 ml wash buffer.
- Combine prepared Ab-conjugated beads with cell lysates.
- Dephosphorylation
- Prepare the CIP mix.
- Add 100 µl CIP mix to the washed beads.
- Incubate at 37 °C and 1,400 rpm in Eppendorf® Thermomixer® R for 10 min.
- Immediately transfer the tubes into ice.
- Wash the beads twice with 1 ml High-salt buffer and twice with 1 ml Wash buffer.
- Prepare the CIP mix.
- 3′-adaptor ligation
- Prepare 3′-ligation mix.
- Add 100 µl 3′-ligation mix to the washed beads.
- Incubate at 16 °C and 1,400 rpm in Eppendorf® Thermomixer® R for overnight.
- Wash the beads twice with 1 ml High-salt buffer and twice with 1ml Wash buffer.
- Prepare 3′-ligation mix.
- RNA 5′-end labeling
- Prepare Hot PNK mix.
- Add 80 µl Hot PNK mix to the washed beads.
- Incubate at 37 °C and 1,400 rpm for 15 min.
- Immediately add 5 µl of 10 mM ATP.
- Incubate at 37 °C and 1,400 rpm in Eppendorf® Thermomixer® R for 5 min.
- Wash the beads twice with 1 ml High-salt buffer and twice with 1 ml Wash buffer.
- Prepare Hot PNK mix.
- NuPAGE and membrane transfer
- The AGO-RNA complexes are eluted from the beads by incubating at 75 °C and 1,400 rpm for 10 min in 30 µl 1.5x NuPAGE LDS Sample Buffer.
- 13 µl of the lysate is mixed with 13 µl of NuPAGE LDS Sample Buffer (4x) and 2 µl of NuPAGE® Sample Reducing Agent (10x), and incubate at 75 °C for 10 min.
- Load the samples on a 4-12% NuPAGE Bis-Tris gel according to the manufacturer′s instructions using 800 ml of 1x MOPS running buffer with NuPAGE® Antioxidant.
- Run the gel for 2 h 30 min at 150 V.
- Autoradiograph the gel for about 5~30 min.
- Mark the size bands in a printed figure.
- Transfer the protein-RNA complexes from the gel to a nitrocellulose membrane for 70 min at 130 V using NuPAGE Transfer buffer according to the manufacturer′s instructions.
- After the transfer, rinse the membrane with D. W., then wrap it with plastic wrap.
- Autoradiograph the gel for about 5~30 min.
- Mark the size bands in a printed figure (Figure 2).
- The AGO-RNA complexes are eluted from the beads by incubating at 75 °C and 1,400 rpm for 10 min in 30 µl 1.5x NuPAGE LDS Sample Buffer.
- Western blotting
- Take out the transferred membrane part for Western blotting and place it into 50-100 ml of 0.1% (v/v) PBS-T with 5% (w/v) BSA.
- Incubate the membrane for 1 h with gentle shaking at room temperature.
- Wash the membrane briefly with 0.1% PBS-T twice.
- Place each membrane into a small boat and pour primary antibody solution (20 ml of 0.1% PBS-T with 5% BSA and 20 µl of anti-AGO mAb 21D2) into the boat.
- Incubate them for at least 4 h with gentle shaking at 4 °C.
- Wash the membrane briefly with 0.1% PBS-T twice.
- Place the membrane into a small boat and pour secondary antibody solution (20 ml of 0.1% PBS-T with 5% BSA and 4 µl of HRP-conjugated Goat Anti-Mouse IgG) into the boat.
- Incubate the membrane for 1 h with gentle shaking at room temperature.
- Wash the membrane with 0.1% PBS-T at least five times for 10 min each with gentle shaking.
- Expose using Chemidoc for ECL detection (Figure 2B).
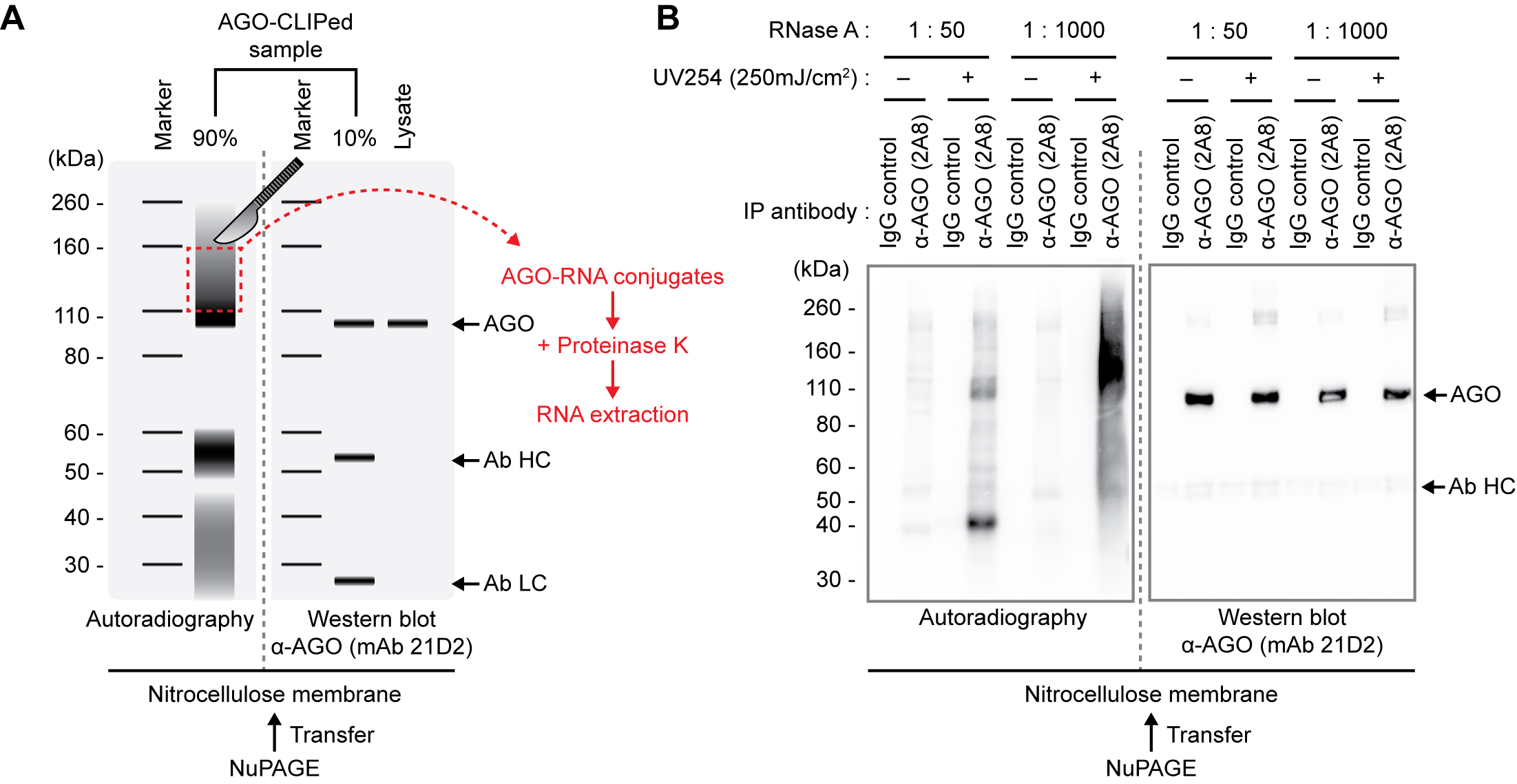
Figure 2. Schematic representation of autoradiography and Western blots of CLIPed AGO-RNA complexes and lysates transferred to nitrocellulose membrane from NuPAGE gel. A. Graphical representation. The region that should be excised by razor and Proteinase K treatment is indicated by thered-dotted rectangle. To evaluate the IP efficiency of AGO-CLIP-seq procedures and the exact unconjugated AGO protein position in the gel, western blotting should be performed in the same gel with autoradiographs. B. Representative data with various conditions of RNase A and UV treatment and antibody used. As viewed, the higher concentration of RNase A is used, the less autoradiographic intensity of AGO-RNA complexes is observed. UV-untreated and using IgG control for IP produces no signal of intact complexes. AGO, Argonaute protein; Ab HC, antibody heavy chain; Ab LC, antibody light chain.
- Take out the transferred membrane part for Western blotting and place it into 50-100 ml of 0.1% (v/v) PBS-T with 5% (w/v) BSA.
- RNA isolation and size fractionation
- Prepare PK mix.
- Cut the piece of 110~160 kDa Band with a razor and place it into a 1.5 ml tube. (Figure 2)
- Add 200 µl PK mix into the membrane-containing tube and incubate at 50 °C and 1,400 rpm for 1 h.
- Add 200 µl PK/Urea buffer and incubate at 50 °C and 1,400 rpm in Eppendorf® Thermomixer® R for 1 h.
- Add 400 µl RNA phenol/chloroform and incubate at 30 °C and 1,400 rpm in Eppendorf® Thermomixer® R for 5 min.
- Centrifuge at 20,000 x g for 5 min at room temperature.
- Transfer the upper (aqueous) phase into a new 1.5 ml tube carefully.
- Add 1 µl linear acrylamide and 40 µl 3 M Sodium acetate pH 5.5 and vortex.
- Add 1 ml of ethanol (EtOH)/isopropanol (1:1), vortex thoroughly and incubate overnight at -20 °C or -80 °C.
- Centrifuge at 4 °C and 28,000 x g for 30 min.
- Wash the pellet once with cold 75% EtOH and subsequently once with 100% EtOH.
- Air-dry the pellet at room temperature for about 10 min.
- Resuspend the pellet with 5 µl Nuclease-free D.W. and add 5 µl 2x RNA Loading buffer.
- Prepare the radiolabeled decade RNA marker using mirVanaTM Probe & Marker Kit.
- Prepare Pre-cast 6% TBE-7M Urea gel.
- Incubate the sample tube at 95 °C for 5 min.
- Load the samples and marker into the prepared gels and run at 200 V until the Bromophenol blue (lower) dye has migrated 75% of the gel length.
- Autoradiograph the gel.
- Cut the gel ranged by 40~77 nt, transfer into a new 1.5 ml tube and crush the gel into small pieces (Figure 3A).
- Add 400 µl 0.3 M Sodium acetate pH 5.5 and rotate overnight at 4 °C.
- Filter the elution supernatant at 4 °C, 20,000 x g for 15 min using a Costar Spin-X centrifugation column.
- Transfer 400 µl of eluate into a new tube.
- Add 1 µl linear acrylamide and 1ml EtOH/isopropanol (1:1), vortex, and incubate for over 1 h at -80 °C.
- Centrifuge at 4 °C and 28,000 x g for 30 min.
- Wash the pellet once with cold 75% EtOH and subsequently once with 100% EtOH.
- Air-dry the pellet at room temperature for about 10 min.

Figure 3. Schematic representation of autoradiographs of 3'-adaptor-labeled CLIPed RNAs (A) and 5'-adaptor/3'-adaptor-labeled AGO-CLIPed RNAs (B). A. Steps of gel excision, RNA extraction from the gel, 5′-adaptor ligation, and phenol extraction of the ligated product are shown; B. Procedure of gel running, gel excision, RNA extraction from the gel, reverse transcription and PCR steps are shown. (A, B)The region that should be excised by razor is indicated by the red-dotted rectangles.
- Prepare PK mix.
- 5′-adaptor ligation and ligate isolation
- Resuspend the pellet in 6.5 µl Nuclease-free D. W. and transfer the resuspended RNA into a new tube.
- Prepare 5′-ligation mix.
- Incubate at 95 °C for 2 min and immediately place on ice (denaturation).
- Add 1 µl T4 RNA ligase and 0.5 µl SUPERase•In RNase Inhibitor for total 20 µl.
- Incubate overnight at 16 °C.
- Add 1 µl linear acrylamide, 20 µl 3 M sodium acetate (pH 5.5), 165 µl Nuclease-free D. W. and 800 µl EtOH:Isopropanol (1:1).
- Vortex thoroughly and incubate for at least 1 h at -80 °C.
- Centrifuge at 4 °C and 28,000 x g for 30 min.
- Wash the pellet once with cold 75% EtOH and subsequently once with 100% EtOH.
- Air-dry the pellet at room temperature for about 10 min.
- Resuspend the pellet with 5 µl Nuclease-free D. W. and add 5 μl 2x RNA Loading buffer.
- Prepare Pre-cast 6% TBE-7M Urea gel and incubate the sample tube at 95 °C for 5 min.
- Load the samples and decade marker into the prepared a pre-cast gel and run at 200 V until the Bromophenol blue (lower) dye has migrated 75% of the gel length.
- Autoradiograph the gel.
- Cut the gel ranged by 65~102 nt, transfer into a new 1.5 ml tube and crush the gel into small pieces (Figure 3B).
- Add 400 µl 0.3 M Sodium acetate pH 5.5 and rotate at 4 h at 4 °C.
- Filter the elution supernatant at 4 °C, 20,000 x g for 15 min using Costar Spin-X centrifugation column.
- Transfer 400 µl of eluate into a new tube.
- Add 1 µl linear acrylamide and 1 ml EtOH/isopropanol (1:1), vortex, and incubate for over 1 h at -80 °C.
- Centrifuge at 4 °C and 28,000 x g for 30 min.
- Wash the pellet once with cold 75% EtOH and subsequently once with 100% EtOH.
- Air-dry the pellet at room temperature for about 10 min.
- Resuspend the pellet in 6.5 µl Nuclease-free D. W. and transfer the resuspended RNA into a new tube.
- Reverse transcription
- Resuspend the pellet in 7 µl nuclease-free D. W. and transfer the resuspended sample into a new tube.
- Mix 7 µl 5′/3′adaptor-ligated AGO-CLIPed RNA with 1 µl 10 μM RNA RT Primer, 5 µl of 2 mM dNTP.
- Heat mixture to 65 °C for 5 min and incubate on ice for at least 1 min.
- Collect the contents of the tube by brief centrifugation.
- Add 4 µl of 5x First-Strand Buffer, 1 µl of 0.1 M DTT, 1 µl of SUPERase•In RNase Inhibitor, 1 µl of SuperScriptTM III RT to total 20 µl.
- Mix by pipetting gently up and down.
- Incubate at 55 °C for 1 h.
- Inactivate the reaction by heating at 70 °C for 15 min.
- Resuspend the pellet in 7 µl nuclease-free D. W. and transfer the resuspended sample into a new tube.
- PCR
- Prepare PCR mix.
- Run PCR reaction with the following thermal program:
- 98 °C, 30 sec
- 98 °C, 10 sec/(3) 55 °C, 30 sec/(4) 72 °C, 10 sec (N cycles)
- 72 °C, 10 min
- 4 °C, ∞
- 98 °C, 30 sec
- Add 1 µl linear acrylamide, 20 µl 3 M sodium acetate (pH 5.5), 130 µl D. W. and 800 µl EtOH:Isopropanol (1:1).
- Vortex thoroughly and incubate for at least 1 h at -80 °C.
- Centrifuge at 4 °C and 28,000 x g for 30 min.
- Wash the pellet once with cold 75% EtOH and subsequently once with 100% EtOH.
- Air-dry the pellet at room temperature for about 10 min.
- Prepare Pre-cast 6% TBE gel.
- Resuspend the pellet in 10 µl D.W. and add 2 µl 6x DNA Loading buffer (with bromophenol blue + xylene cyanol).
- Load the sample and Low Molecular Weight DNA Ladder into prepared gel and run at 100 V until bromophenol blue (lower) dye reaches the bottom.
- Stain the gel in EtBr-containing 1x TBE by gentle rocking for 10 min.
- Identify and cut the gel of 136~173 bp amplicon (Figure 4). It should contain 18~55 nt CLIPed inserts flanked by 5′- and 3′-primer sequences. And generally, if the quantity of substrates for PCR is enough, it can be visualized clearly at 20 cycles in a size of 136~173 bp region in the gel.
- Add 400 µl 0.3 M Sodium acetate pH 5.5 and rotate overnight at 4 °C.
- Filter the elution supernatant at 4 °C, 20,000 x g for 15 min using Costar Spin-X centrifugation column.
- Transfer 400 µl of eluate into a new tube.
- Add 1 µl Linear acrylamide and 1ml EtOH/isopropanol (1:1), vortex, and incubate for over 1 h at -80 °C.
- Centrifuge at 4 °C and 28,000 x g for 30 min.
- Wash the pellet once with cold 75% EtOH and subsequently once with 100% EtOH.
- Air-dry the pellet at room temperature for about 10 min.
- Resuspend the pellet with an appropriate volume of water (~20 µl).
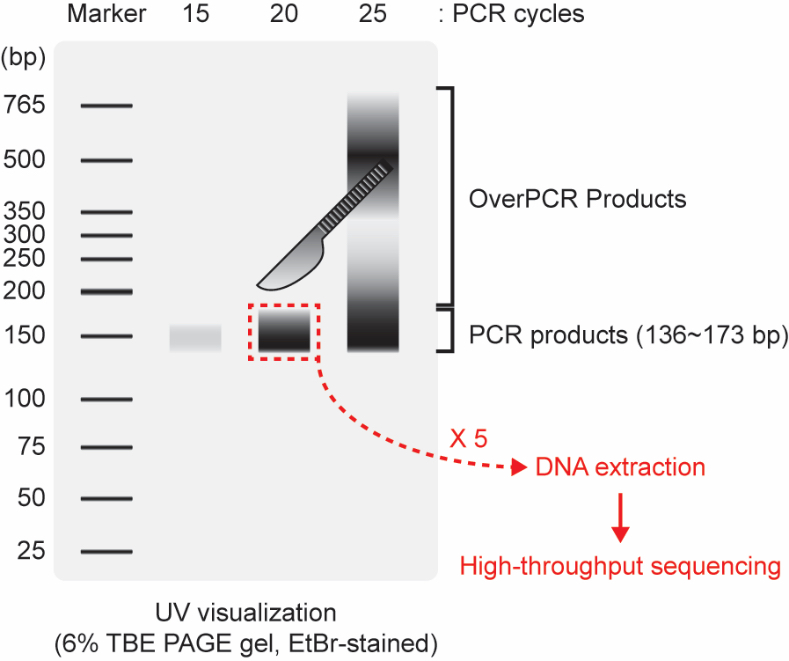
Figure 4. Visualization of PCR products. The region that should be excised by razor is indicated by the red-dotted rectangle. Over-amplified PCR (Over PCR) products are frequently found in >22 PCR cycles if AGO-CLIP-seq is well-performed. We recommend collecting at least five PCR reactions to gather a large enough quantity for library construction and high-throughput sequencing.
- Prepare PCR mix.
- Cell culture and HCMV infection
- Library construction for mRNA-seq and smallRNA-seq
- Library for mRNA-seq
Note: The mRNAseq libraries can be prepared as previously described (Guo et al., 2010) with the following modifications.- With one of five dishes (from step A2b), extract total RNA using Trizol® reagent according to manufacturer’s instruction.
- Enrich poly(A)+ RNAs using Dynabeads® mRNA DIRECT™ Purification Kit according to manufacturer’s instruction with 5 μg of total RNA.
- 0.2 μg of oligo (dT)-enriched RNAs are fragmented using the NEBnext Magnesium RNA Fragmentation module.
- Dephosphorylate the fragmented RNAs using Antarctic phosphatase at 37 °C for 1 h and heat-inactivate at 70 °C for 5 min.
- Label the 5'-end of dephosphorylated RNAs using [γ-32P] ATP and PNK.
- Labeled 35–55-nt RNA fragments are purified from a 10% TBE-Urea Gel.
- Ligate 3'-adaptor and gel-purify the ligated products.
- Ligate 5'-adaptor and gel-purify the ligated products.
- Perform reverse transcription (RT) and PCR.
- With one of five dishes (from step A2b), extract total RNA using Trizol® reagent according to manufacturer’s instruction.
- Library for smallRNA-seq:
- The smallRNA-seq libraries are prepared using the method described for mRNA-seq libraries except for differences in the initial RNA harvest steps: 5 μg of total RNAs are electrophoresed in a 10% TBE-Urea Gel, and 18–25-nt RNA species are purified for further steps.
- Library for mRNA-seq
- High-throughput sequencing
Amplified cDNA libraries are sequenced by using Illumina HiSeq2000 for 1 x 51 cycles at single read/quadruplexed. - Data processing and bioinformatics
- Preprocessing:
- Sequencing reads are preprocessed before reads alignment by using CutAdap (v. 1.2.1) to remove adapter sequences, fastx_artifacts_filter in FASTX-Toolkit (v. 0.0.13.2, http://hannonlab.cshl.edu/fastx_toolkit/) to filter artifact reads, and then remove low quality read sequences. The options used in CutAdap software are "-m 17 --match-read-wildcards -O 10 -e 0.10".
- To calculate the relative fraction of reads mapped to human rRNA or tRNA, apply Bowtie2 (v. 2.1.0) aligning software to the rRNA reference sequences from Rfam (http://rfam.samger.ac.ur/) (Burge et al., 2013) and the tRNA reference sequences from GtRNAdb (http://gtrnadb.ucsc.edu/) (Chan and Lowe, 2009).
- To be as comprehensive as possible, expand these sequence sets to include homologous sequences found in the human genome (hg19) with BLAST+ (v. 2.2.27) software (Camacho et al., 2009).
- Sequencing reads are preprocessed before reads alignment by using CutAdap (v. 1.2.1) to remove adapter sequences, fastx_artifacts_filter in FASTX-Toolkit (v. 0.0.13.2, http://hannonlab.cshl.edu/fastx_toolkit/) to filter artifact reads, and then remove low quality read sequences. The options used in CutAdap software are "-m 17 --match-read-wildcards -O 10 -e 0.10".
- Genomes and databases:
- All alignments and analyses are conducted based on human hg19 reference sequences and the HCMV Towne genome.
- The genome annotations for genes, lincRNAs, miscRNAs, mt tRNAs, snoRNAs, and snRNAs are downloaded from the UCSC genome table browser and the BioMart in Ensembl for human and NCBI for HCMV, respectively. The miRNA annotations of human and HCMV miRNAs are downloaded from miRBase21 (http://mirbase.org/).
- Because most of HCMV-encoded miRNAs are annotated by studies using other kinds of HCMV strains, their annotated locations are not concordant to Towne genome.Blast searching is conducted with miRNAs whose annotation is based on AD169 genome to HCMV Towne and miRNAs to obtain the annotation of HCMV miRNA annotations (Stark et al., 2012).
- All alignments and analyses are conducted based on human hg19 reference sequences and the HCMV Towne genome.
- Gene expression measurement:
- Calculate the expression levels of mRNAs and miRNAs using the Tuxedo suite.
- To determine the gene expression level, apply Tophat2 (v. 2.0.9) with "--no-coverage-search --b2-very-sensitive --no-nevel-junc" options and cufflinks (v. 2.1.1) with "-q --no-update-check" options for the calculation of RPKM (reads per kilo base of transcript per million fragments mapped) scores.
- Apply Bowtie2 (v. 2.1.0) with "-k 101, --score-min C,0,0 (no mismatch) or --score-min C,-8,0 (for 1 mismatch) -mp 8,8 --np 8 --very-sensitive" options for the determination of miRNA expression levels with up to one mismatch to calculate RPM (reads per million reads mapped) scores.
- Quantify expression levels of miRNAs from AGO-CLIP-seq data similarly to smallRNA sequencing data. An additional mismatch (--score-min C, -16,0 option in Bowtie2) for AGO-CLIP-seq alignments can be allowed, because AGO-CLIP-seq is more error-prone.
- Calculate the expression levels of mRNAs and miRNAs using the Tuxedo suite.
- AGO binding site detection:
- As AGO-CLIP-seq reads indicate AGO-RNA interactions, a cluster of AGO-CLIP-seq reads (“peak”) suggests an AGO binding site.
- To exclude random AGO-RNA interactions, a peak detection method is devised to measure significant enrichment of AGO-CLIP-seq reads compared to mRNA-seq reads using Fisher's exact test.
Note: This test compares the expression level of a given 60-bp and 1-Mbps local background region in AGO-CLIP-seq and mRNA-seq, respectively. - The resulting P-value is used to define a peak region and to evaluate the significance of the peak.
- Read preparation and alignment
1.)Non-miRNA reads are collected and filtered from AGO-CLIP-seq data.
2.)Align AGO-CLIP-seq reads and mRNA-seq reads onto the human genome using TopHat2 with a less stringent mode (added --b2-score-min L, -0.6, 0.9 in mRNAseq options) to allow for slightly more sequencing errors in AGO-CLIP-seq.
3.)For each genomic position, calculate the aligned reads for AGO-CLIP-seq and mRNA-seq to identifying the peak region.
4.)Since mRNA sequencing has a coverage bias across the transcript, the average over the transcript length is taken for each transcript to identify peaks. - Peak detection
1.)To detect significant peaks from AGO-CLIP-seq data, apply Fisher's exact test using mapped reads in 60-bp candidate peak regions and in the surrounding 1-Mbp background region.
2.)Window size is determined by the AGO footprint spans about 60 bp (Chi et al., 2009), and the background was set to be large enough.
3.)Adjust p-values using Q-VALUE (v. 1.1) from the Storey lab (http://genomics.princeton.edu/storeylab/qvalue) with default settings for multiple comparison correction.
4.)Define peaks with false discovery rates (FDRs) less than 0.01 as significant.
5.)Combine overlapping peaks in a merged peak site to avoid redundancy.
6.)Use peaks located in human exon regions for the analysis of AGO binding sites on human mRNAs. - Increased CLIPseq constraint:
1.)To detect target sites of a specific miRNA on 3'-UTR in mRNAs, examine whether a peak site includes a seed pairing site with canonical seeds sites (8mer, 7mer-A1, 7merm8, and 6mer) of both human and viral miRNAs by searching in the defined peak region (60 bp, longer for merged peaks). Based on this search, peaks in case that there is no seed pairing site in its region can be excluded.
2.)To identify more robust targets of viral miRNA, “Increased CLIP-seq” constraint is applied, representing as the level of enriched AGO-CLIP-seq reads relative to the previous time of viral infection.
3.)To measure the enrichment score for a given microRNA target site, we introduce a relative score called, the “ACE-score (AGO-CLIP-seq Enrichment score)”. ACE-score is defined as the Log ratio of normalized counts at a given time point to the normalized counts at the previous time after viral infection (Figure 5).
Figure 5. Scheme of ACE-score calculation. Formula of ACE-scoring and examples of peak cluster sites are illustrated and adapted from Reference (Kim et al., 2015). RPKM, reads per kilo base of transcript per million fragments mapped.
- Read preparation and alignment
- As AGO-CLIP-seq reads indicate AGO-RNA interactions, a cluster of AGO-CLIP-seq reads (“peak”) suggests an AGO binding site.
- Preprocessing:
Recipes
- Phosphate-buffered saline (PBS)
137 mM NaCl 2.7 mM KCl 10 mM Na2HPO4 2 mM KH2PO4 - Lysis buffer
50 mM Tris-HCl (pH 7.4) 100 mM NaCl 1% NP-40 0.1% SDS 0.5% sodium deoxycholate - High-salt buffer
50 mM Tris-HCl (pH 7.4) 1 M NaCl 1 mM EDTA 1% NP-40 0.1% SDS 0.5% sodium deoxycholate - Wash buffer
20 mM Tris-HCl (pH 7.4) 10 mM MgCl2 0.2% Tween-20 - CIP mix
10 µl 10x NE buffer 3 5 µl Alkaline Phosphatase, Calf Intestinal (CIP) 3 µl SUPERase.In RNase Inhibitor 82 µl nuclease-free distilled water (D. W.) to 100 µl final volume - 3'-ligation mix
100 pmol 3′-Adaptor (RA3) (1 µl 100 µM adaptor) 10 µl 10x T4 RNA ligase buffer (no ATP) 3 µl T4 RNA ligase truncated K227Q 3 µl SUPERase•In RNase Inhibitor 30 µl 50% PEG8000 53 µl Nuclease-free D. W. to 100 µl final volume - Hot PNK mix
5 µl 10 mCi/ml [γ-32P] ATP 8 µl 10x T4 PNK buffer 3 µl T4 PNK buffer 2 µl SUPERase.In RNase Inhibitor 62 µl Nuclease-free D. W. to 80 µl final volume - 0.1% PBS-T
137 mM NaCl 2.7 mM KCl 10 mM Na2HPO4 2 mM KH2PO4 0.1% (v/v) Tween-20 - PK buffer
100 mM Tris-HCl (pH 7.4) 50 mM NaCl 10 mM EDTA - PK mix
100 µl PK buffer 100 µl 20 mg/ml Proteinase K - PK/Urea buffer
100 mM Tris-HCl (pH 7.4) 50 mM NaCl 10 mM EDTA 7 M Urea - 5'-ligation mix
8.5 µl Resuspended sample 1 µl 5′-adaptor oligonucleotide 2 µl 10x T4 RNA ligase buffer (with ATP) 6 µl 50% PEG 8000 (NEB) 1 µl 0.1% BSA (supplied in TaKaRa T4 RNA ligase kit) - PCR mix
2 µl RT product (template) 2 µl 5′-Primer (10 µM) 2 µl 3′-Primer (10 µM) 10 µl 5x HF buffer 5 µl 2 mM dNTPs mix 0.5 µl phusion polymerase 28.5 µl D. W. to total 50 µl - 6x DNA loading buffer
10 mM Tris-HCl (pH 7.6) 0.03% bromophenol blue (w/v) 0.03% Xylene cyanol FF (w/v) 60% glycerol 60 mM EDTA - TBE (Tris-borate-EDTA)
- Prepare a 5x stock solution in 1 L of D. W.:
54 g Tris base 27.5 g boric acid 20 ml 0.5 M EDTA (pH 8.0) - The 0.5x working solution is 45 mM Tris-borate/1 mM EDTA.
- Prepare a 5x stock solution in 1 L of D. W.:
Acknowledgments
We want to thank the Group of Daehyun Baek (School of Biological Sciences, Seoul National University, Seoul, Republic of Korea) for the development and technical support of bioinformatic analysis. This protocol has been adapted from our previous study (Kim et al., 2015). This work was supported by IBS-R008-D1 of Institute for Basic Science from the Ministry of Science, ICT and Future Planning of Korea (K. A.).
References
- Burge, S. W., Daub, J., Eberhardt, R., Tate, J., Barquist, L., Nawrocki, E. P., Eddy, S. R., Gardner, P. P. and Bateman, A. (2013). Rfam 11.0: 10 years of RNA families. Nucleic Acids Res 41(Database issue): D226-232.
- Britt, W. J. (2010). Human cytomegalovirus: propagation, quantification, and storage. Curr Protoc Microbiol Chapter 14: Unit 14E 13.
- Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K. and Madden, T. L. (2009). BLAST+: architecture and applications. BMC Bioinformatics 10: 421.
- Chan, P. P. and Lowe, T. M. (2009). GtRNAdb: a database of transfer RNA genes detected in genomic sequence. Nucleic Acids Res 37(Database issue): D93-97.
- Chi, S. W., Zang, J. B., Mele, A. and Darnell, R. B. (2009). Argonaute HITS-CLIP decodes microRNA-mRNA interaction maps. Nature 460(7254): 479-486.
- Guo, H., Ingolia, N. T., Weissman, J. S. and Bartel, D. P. (2010). Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature 466(7308): 835-840.
- Kim, S., Seo, D., Kim, D., Hong, Y., Chang, H., Baek, D., Kim, V. N., Lee, S. and Ahn, K. (2015). Temporal landscape of microRNA-mediated host-virus crosstalk during productive human cytomegalovirus infection. Cell Host Microbe 17(6): 838-851.
- Stark, T. J., Arnold, J. D., Spector, D. H. and Yeo, G. W. (2012). High-resolution profiling and analysis of viral and host small RNAs during human cytomegalovirus infection. J Virol 86(1): 226-235.
Article Information
Copyright
© 2016 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Kim, S. and Ahn, K. (2016). ACE-score-based Analysis of Temporal miRNA Targetomes During Human Cytomegalovirus Infection Using AGO-CLIP-seq . Bio-protocol 6(8): e1791. DOI: 10.21769/BioProtoc.1791.
Category
Microbiology > Microbial biochemistry > RNA
Biochemistry > RNA > miRNA targeting
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.