- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Structured Illumination Microscopy (SIM) and Photoactivated Localization Microscopy (PALM) to Analyze the Abundance and Distribution of RNA Polymerase II Molecules on Flow-sorted Arabidopsis Nuclei

Published: Vol 6, Iss 3, Feb 5, 2016 DOI: 10.21769/BioProtoc.1725 Views: 13697
Reviewed by: Samik BhattacharyaAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Mar 2015
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
RNA polymerase II (RNAPII) is the enzyme transcribing most of the eukaryotic protein-coding genes. Analysing the distribution and quantification of RNAPII can help understanding its function in interphase nuclei. Although several investigations in mammals indicate the organization of RNAPII in so-called ‘transcription factories’ (Jackson et al., 1993; Rieder et al., 2012; Papantonis and Cook, 2013), their existence is still controversially discussed (Zhao et al., 2014).
Recently, based on super-resolution microscopy the presence of transcription factories was also suggested in plants. Applying structured illumination microscopy (SIM) and photoactivated localization microscopy (PALM) the distribution and number of RNAPII molecules in Arabidopsis nuclei were analysed and a positive correlation between RNAPII abundance and endopolyploidy was found (Schubert, 2014; Schubert and Weisshart, 2015).
Here, we present a protocol describing the isolation of Arabidopsis thaliana interphase nuclei via flow-sorting according to their endopolyploidy level, followed by a double immunostaining using antibodies specific for different RNAPII modifications and the subsequent evaluation by spatial SIM and PALM to achieve results regarding the abundance, distribution and co-localization of single inactive and active RNAPII molecules.
Part I. Flow sorting of Arabidopsis interphase nuclei
Materials and Reagents
- 22x22 mm high precision coverslips (170±5 µm, no. 1.5H) (Marienfeld-Superior) Rubber cement (Marabu Australia, catalog number: 290110000 )
- 5 ml Polystyrene Round-Bottom Tubes with 35 µm cell strainer cap (BD Biosciences, catalog number: 352235 ) or alternatively, 5 ml Polystyrene Round-Bottom Tube (BD Biosciences, catalog number: 352063 ) in combination with disposable filters CellTrics, 30 or 50 µm (Sysmex-Partec, catalog numbers: 04-0042-2316 or 04-0042-2317 )
Note: Currently, it is "Corning, Falcon®, catalog number: 352235” and "Corning, Falcon®, catalog number: 352063”. - 0.22 µm filter
- A. thaliana rosette leaves
- Calibration beads [SpheroTM Rainbow Calibration Particles (8 peaks)] (BD Biosciences, catalog number: 559123 )
- Accudrop Beads (BD FACSTM Accudrop Beads) (BD Biosciences, catalog number: 345249 )
- Formaldehyde solution 37% (Carl Roth GmbH + Co, catalog number: 7398.1 )
- 4', 6-diamidino-2-phenylindole (DAPI) (Life Technologies, Molecular ProbesTM, catalog number: D-1306 )
Note: Currently, it is “Thermo Fisher Scientific, Molecular ProbesTM, catalog number: D-1306”. - Milli-Q water
- NaCl
- Na2HPO4.2H2O
- NaH2PO4
- Tris
- Na2EDTA
- Spermin
- KCl
- Triton X-100
- NaOH
- β-mercaptoethanol
- MgCl2
- Sucrose
- Tween 20
- HCl
- BD FACSFlow sheath fluid (BD Biosciences, catalog number: 342003 ) or alternatively, 1 x PBS (see Recipes)
- Formaldehyde fixative (see Recipes)
- 500 ml Tris-Buffer (see Recipes)
- 200 ml lysis buffer LB01 (Dolezel et al., 1989) (see Recipes)
- 100 µg/ml DAPI stock solution (see Recipes)
- 25 ml sucrose buffer (see Recipes)
Equipment
- FACSAria IIu (BD Biosciences)
Software
- FACSDiva software
Procedure
- Nuclei isolation
- Harvest 2-3 rosette leaves of four to six weeks old plants of A. thaliana into distilled (or deionized) water.
- Fix the plant tissue by transferring it into the formaldehyde fixative and incubate it for 20 min in a small glass beaker on ice under vacuum using a desiccator.
- Dry off the plant material shortly on filter paper.
- Wash in Tris-buffer 2x 10 min in a new glass beaker on ice, and dry off the plant material shortly on filter paper between the washing steps.
- Transfer the plant tissue in a pre-cooled Petri dish, add roughly 250 µl of LB01 lysis buffer and chop it with a sharp razor blade until getting a fine suspension.
- Add additional 250-750 µl (depending on the amount of used plant tissue) of LB01 lysis buffer and filter the suspension through a 30-50 µm mesh (using either tubes with cell strainer caps or other disposable filters) into a 5 ml Round-Bottom polystyrene tube and store it on ice. This suspension can either be used directly to prepare nuclei preparations suitable for immunostaining experiments (continue with procedure C ‘Preparation of coverslips’) or used for fluorescence activated cell sorting (FACS) before. The latter variant allows the separation of nuclei according to different ploidy levels and results in preparations of high purity that guarantee a minimum of background.
- Stain the suspension by adding DAPI stock solution to a final concentration of 1-2 µg/ml and store it for 10 min on ice.
- Harvest 2-3 rosette leaves of four to six weeks old plants of A. thaliana into distilled (or deionized) water.
- Sorting of nuclei using a FACSAria cell sorter
- Switch on the computer, the cytometer and the blue (488 nm) and violet (405 nm) or (Near-) UV (375 nm) laser and wait 30 min for them to warm up. Start the FACSDiva software, check the level of the fluidics in the cytometer window and refill sorting buffer (BD FACSFlow or 1x PBS) and empty the waste if needed. Perform fluidics startup, insert a 70 µm nozzle and switch on the stream. Control if the correct sort setup is selected (‘Sort’ - ‘Sort Setup’ - ’70 micron’). Switch on ‘Sweet Spot’ in the ‘Breakoff’ window.
- Run calibration beads to control the performance of the flow sorter. If 8 peaks are not displayed in corresponding log scale histograms for FITC and DAPI, clean the flow cell according to the FACSAria User’s Guide or the instructions of the operator.
- Run Accudrop Beads and determine the optimal drop delay using the ‘Auto Drop Delay’ function of the FACSAria according to FACSAria User’s Guide.
- Run nuclei sample, set up the trigger to DAPI fluorescence and select a proper threshold level that allows you to display the signals on a dot plot of side scatter (SSC) area (log scale) versus DAPI fluorescence area (log scale). Adjust the voltage of the photomultiplier for the DAPI fluorescence to achieve a displaying of all ploidy level populations (Figure 1A).
- Define a gate around the nuclei populations and select this to be displayed in a histogram of DAPI fluorescence area. Define sorting regions for the ploidy levels of interest (Figure 1B).
- Open the ‘Sort Layout’ window, select collection device ‘4 Tube’, precision mode ‘Purity’ and define the sort locations. Insert empty Eppendorf tubes into the collection device holders.
- Sort the required number of nuclei (10,000 to 50,000; depending on the availability of the individual ploidy levels and the desired number of coverslips) into collection tubes and store them afterwards on ice.
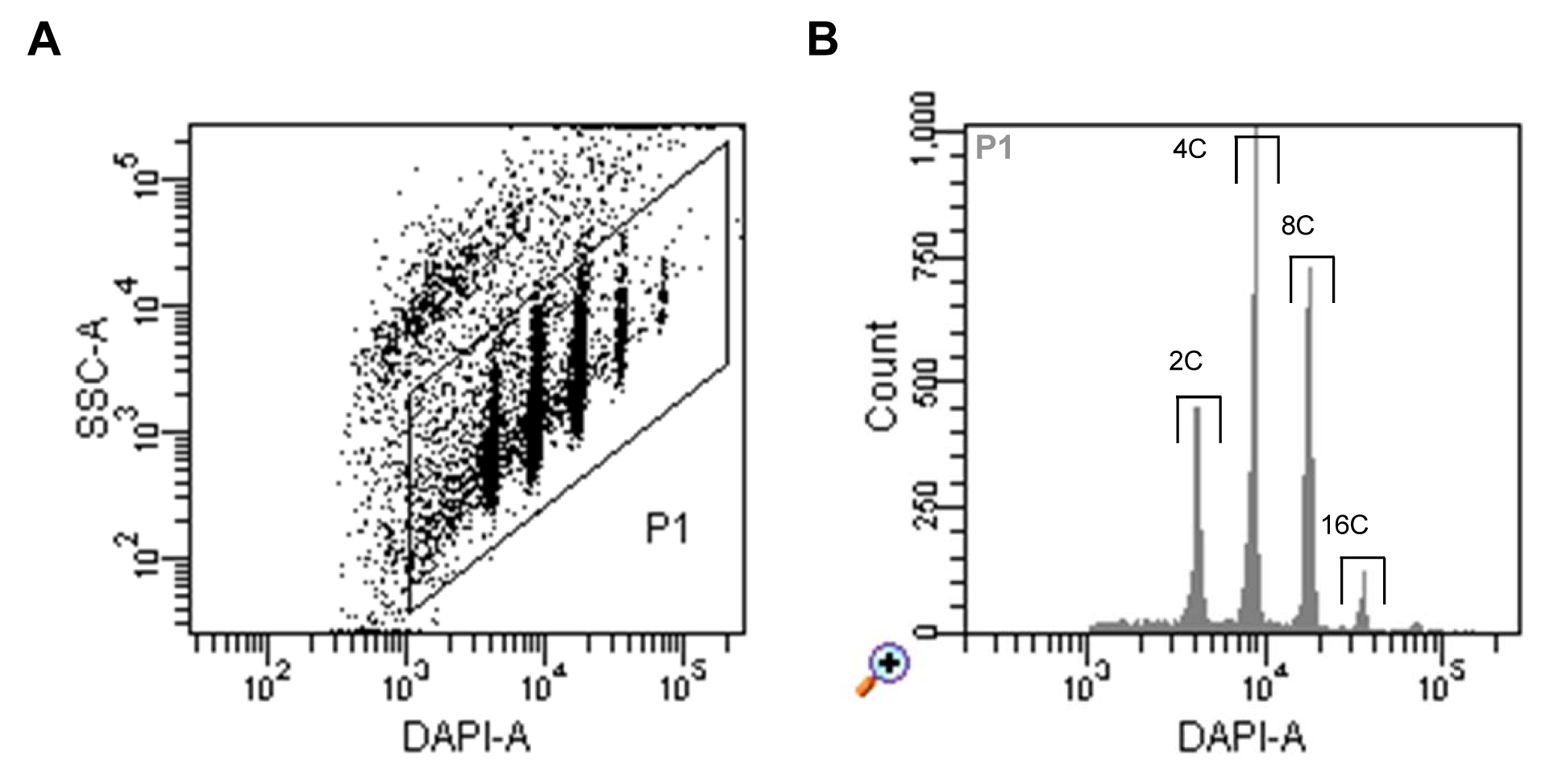
Figure 1. Flow sorting of A. thaliana interphase nuclei. A. Dot plot of SSC area versus DAPI fluorescence area indicating populations of nuclei with different DAPI fluorescence intensities reflecting different levels of endopolyploidy. B. Histogram of DAPI fluorescence of particles gated as P1 in (A). The gates above of the individual peaks indicate the sorting gates of 2C, 4C, 8C and 16C nuclei.
- Switch on the computer, the cytometer and the blue (488 nm) and violet (405 nm) or (Near-) UV (375 nm) laser and wait 30 min for them to warm up. Start the FACSDiva software, check the level of the fluidics in the cytometer window and refill sorting buffer (BD FACSFlow or 1x PBS) and empty the waste if needed. Perform fluidics startup, insert a 70 µm nozzle and switch on the stream. Control if the correct sort setup is selected (‘Sort’ - ‘Sort Setup’ - ’70 micron’). Switch on ‘Sweet Spot’ in the ‘Breakoff’ window.
- Preparation of coverslips
- Pipette 12 to 15 µl of sucrose buffer onto 22x22 mm high precision coverslips (Marienfeld) attached on slides by rubber cement (Marabu) (Figure 2A).
- Subsequently pipette equivalent volumes of sorted nuclei into the drop of sucrose buffer. Gently mix the solutions with a pipette tip.
- Air-dry coverslips over-night at room temperature.
- Coverslips can be used either immediately for immunostaining or stored for up to several months at -20 °C in the dark.
- Pipette 12 to 15 µl of sucrose buffer onto 22x22 mm high precision coverslips (Marienfeld) attached on slides by rubber cement (Marabu) (Figure 2A).
Recipes
- 1 L 1x PBS
154 mM NaCl: 9 g
7 mM Na2HPO4.2H2O: 1.246 g
4 mM NaH2PO4: 0.48 g
Dilute in A.dest. and adjust the pH to 7.0 using 1 N NaOH - Formaldehyde fixative
4% formaldehyde in Tris buffer (dilute 6 ml formaldehyde solution 37%, with 50 ml Tris buffer), the fixative should be prepared just before use. Attend to work with formaldehyde under a hood and dispose waste properly. - 500 ml Tris-buffer
10 mM Tris: 0.605 g
10 mM Na2EDTA: 1.861 g
100 mM NaCl: 2.922 g
0.1% Triton X-100: 500 µl
Dilute in A.dest. and adjust the pH to 7.5 using 1 N NaOH - 200 ml Lysis buffer LB01 (Dolezel et al., 1989)
15 mM Tris: 363 mg
2 mM Na2EDTA: 148.9 mg
0.5 mM Spermin: 20.2 mg
80 mM KCl: 1.193 g
20 mM NaCl: 233.8 mg
0.1% Triton X-100: 200 µl
Adjust pH to 7.5 using 1 N NaOH and filter through a 0.22 µm filter to remove small particles. Add 220 µl β–mercaptoethanol and store in 10 ml aliquots at -20 °C. Attend to work with β-mercaptoethanol under a hood and dispose waste properly. - 100 µg/ml DAPI stock solution
Dissolve 5 mg 4', 6-diamidino-2-phenylindole (DAPI) in 50 ml deionized water by stirring for 60 min and filter through a 0.22 µm filter to remove small particles. Store in 0.5 ml aliquots at -20 °C. - 25 ml Sucrose buffer
10 mM Tris: 30.3 mg
50 mM KCl: 93.2 mg
2 mM MgCl2·6H2O: 10.2 mg
5% sucrose: 1.25g
0.05% Tween 20: 12.5 µl
Adjust pH to 8.0 using 5 N HCl and store in 1 ml aliquots at -20 °C
Figure 2. Specimen preparation, immunostaining procedure and coverslip handling for microscopy. A. Fixing the coverslip at a slide by applying rubber cement; B. Adding of the blocking solution; C-D. Covering with parafilm. E. Slide placed in a humidity chamber; F. Cutting the rubber cement with a razor blade; G. Lifting the coverslip; H. Pipetting 1% 2-mercaptoethanol in 1x PBS into the coverslip containing coverslip chamber.
Part II. Immunolabeling of sorted nuclei
Materials and Reagents
- Primary antibodies
- Secondary antibodies
- Goat anti-mouse-Cy5 (1:300) (Jackson ImmunoResearch Inc., catalog number: 715-175-151 )
- Goat anti-rat Alexa488 (1:200) (Jackson ImmunoResearch Inc., catalog number: 112-545-167 )
- Goat anti-mouse-Cy5 (1:300) (Jackson ImmunoResearch Inc., catalog number: 715-175-151 )
- Milli-Q water
- PIPES
- MgSO4
- EGTA (pH 6.9)
- BSA
- Triton X-100
- 1x MTSB (microtubule stabilizing) buffer (see Recipes)
- Blocking solution (see Recipes)
- Dilution buffer for antibodies (see Recipes)
Equipment
- Humidity chamber
- Slide cuvettes [e.g. vertical glass cuvettes (Electron Microscopy Sciences, catalog number: 70318-04 )]
Procedure
- After storage at -20 °C keep coverslips (fixed on slides by rubber cement) 10 min at room temperature (RT); then, place them in suitable racks and dry in an incubator at 60 °C for 45 min.
- Wash 5 min in 1x MTSB buffer in a slide cuvette.
- Add 60 µl of blocking solution to each sample, cover with parafilm and incubate 1 h at RT in a humidity chamber (Figures 2B-E).
- Dilute the primary antibodies in dilution buffer.
- Remove the parafilm and blocking solution, then add 40 µl of the primary antibody solution to each coverslip, cover with parafilm and incubate overnight at 10 °C in a humidity chamber.
- Wash the coverslips 3x 5 min in 1x MTSB buffer by placing the slide into a cuvette.
- Dilute the secondary antibodies, add 40 µl of this dilution to each coverslip, cover with parafilm and incubate overnight at 10 °C in a humidity chamber.
- Wash the coverslips 1x 5 min in 1x MTSB buffer in a slide cuvette.
- Dehydrate in ethanol series (70%, 90%, 96%) for 5 min each.
- Air dry in dark.
Recipes
- 1x MTSB (microtubule stabilizing buffer)
50 mM PIPES
5 mM MgSO4
5 mM EGTA (pH 6.9) - Blocking solution
8% BSA
0.1% Triton X-100 in 1x MTSB - Dilution buffer for antibodies
1% BSA in 1x MTSB buffer
Store in 1 ml aliquots at -20 °C.
Part III. SIM and PALM evaluation
This protocol is based on using the ELYRA PS.1 system with the software ZEN 2012. Systems from other suppliers have similar functions. Therefore, the protocol can well serve also for other commercial systems including the processing steps. Being trained on such systems the resemblance of specific instructions to those given here should become obvious.
Materials and Reagents
- 200 nm TetraSpeck™ Fluorescent Microspheres slide (Carl Zeiss Microscopy GmbH)
Note: TetraSpeck™ Fluorescent Microspheres is a trademark of Thermo Fisher Scientific. - 40 nm FluoSpheres® Carboxylate-Modified Microspheres slide (Carl Zeiss Microscopy GmbH, catalog number: 1783-456 )
- 40 nm FluoSpheres® Carboxylate-Modified Microspheres (Thermo Fisher Scientific)
- 200 nm TetraSpeckTM microspheres (Thermo Fisher Scientific, catalog number: T-7280 )
- 2-mercaptoethanol (Sigma-Aldrich, catalog number: 63689 )
- NaCl
- Na2HPO4.2H2O
- NaH2PO4
- NaOH
- 1x Phosphate-buffered saline (PBS) (see Recipes)
- Phosphate buffered saline solution 10x concentrate (10x PBS) (Sigma-Aldrich, catalog number: P5493 ) (see Recipes)
Equipment
- Coverslip chamber 'Chamlide' for 22x22 mm coverslips (Live Cell Instruments, catalogue number: CM-S22-1 )
- Microscope system Elyra PS.1 (Carl Zeiss Microscopy GmbH)
Software
- Software ZEN 2012 (Carl Zeiss Microscopy GmbH)
Procedure
- Sample mounting and nuclei selection
- Remove the coverslip from the slide by carefully cutting the rubber cement with a razor blade at one side and lifting it from the slide (Figures 2F-G). Place the coverslip into a 22x22 mm coverslip chamber and cover with 1% 2-mercaptoethanol in 1x PBS (Figure 2H). Close with the lid.
- Select favorable nuclei using the 63x/1.4 Oil Plan-Apochromat objective, X-Cite (LED) illumination, the eyepieces and apply the ZEN tool 'Locate' (Figure 3).
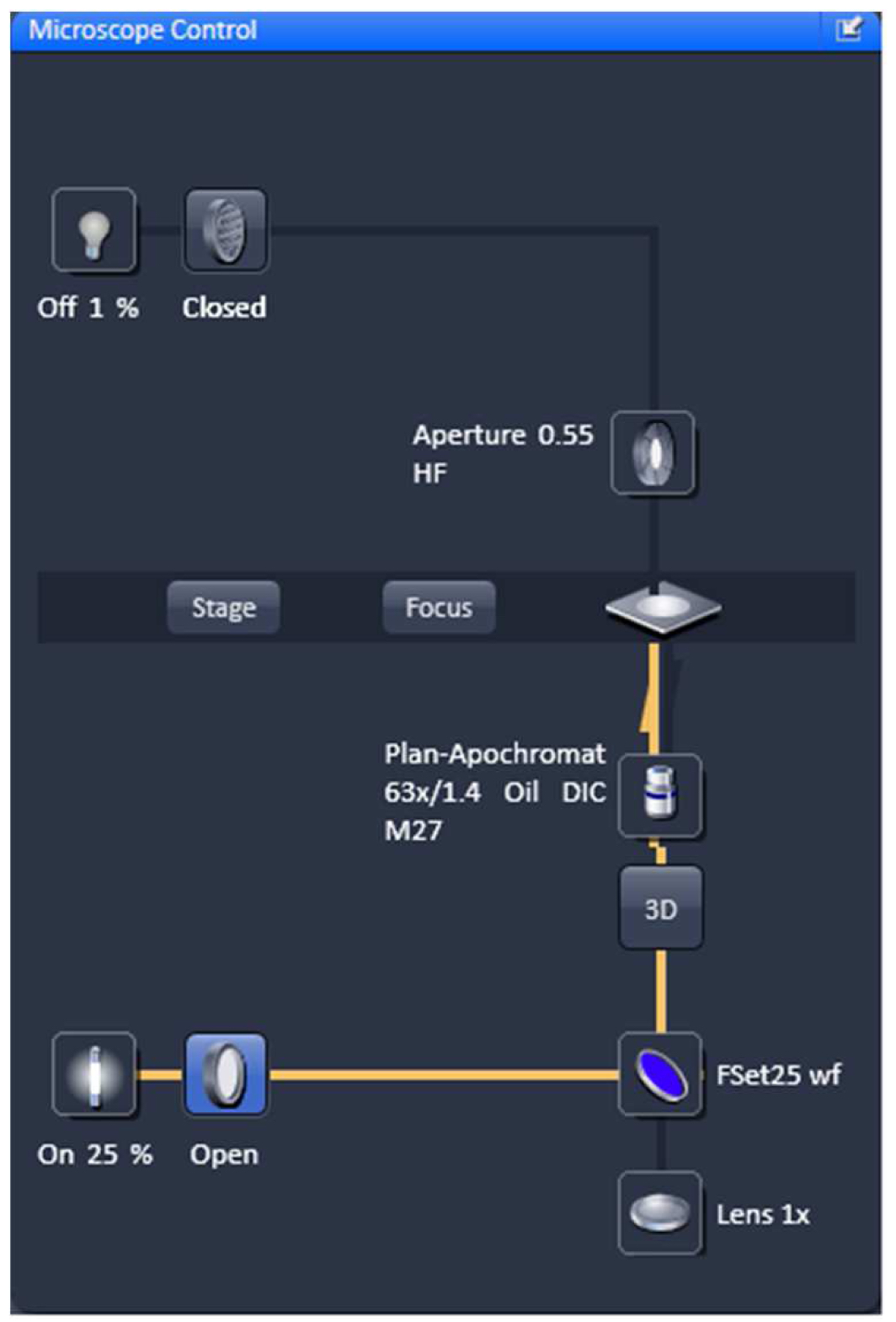
Figure 3. Microscope control by 'Locate'. Settings for reflected fluorescence light ocular detection are activated (yellow line).
- Remove the coverslip from the slide by carefully cutting the rubber cement with a razor blade at one side and lifting it from the slide (Figures 2F-G). Place the coverslip into a 22x22 mm coverslip chamber and cover with 1% 2-mercaptoethanol in 1x PBS (Figure 2H). Close with the lid.
- Raw data acquisition for 3D SIM
Using the same objective, first acquire the SIM image and then the PALM image (as described in the next section) by exciting with the 642 nm and 488 nm lasers subsequently (51 µm grid for 642 nm and 34 µm grid for 488 nm excitations; 5 rotations). Starting with the longer wavelength minimizes bleaching.- Switch from tool 'Locate' to the tool 'Acquisition' and select in the ‘Imaging Setup’ tool ‘SIM’ to acquire SIM raw data image stacks (Figure 4).
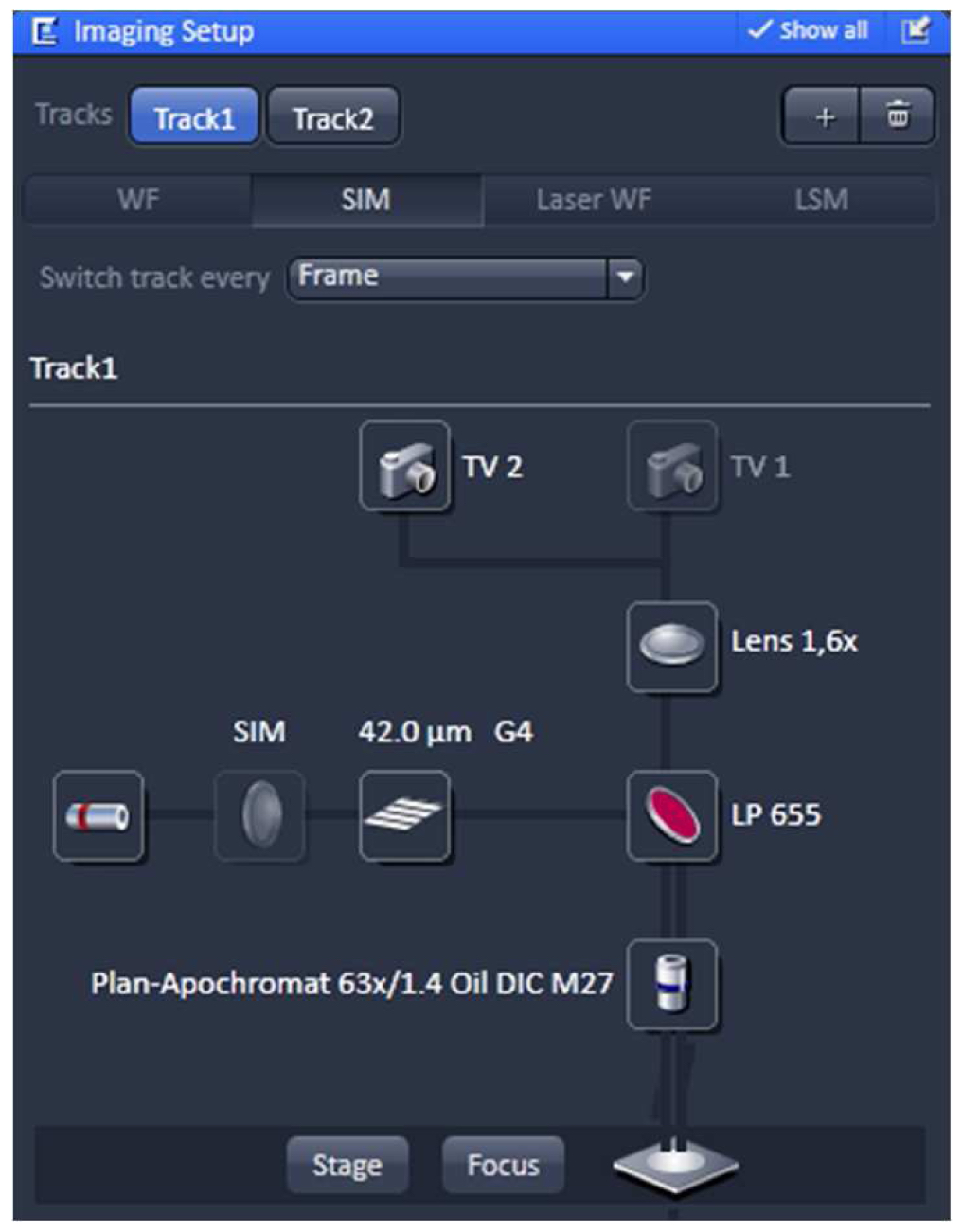
Figure 4. ‘Imaging Setup’ tool of ZEN ‘Acquisition’ for SIM. ‘SIM’ is selected and a two track experiment with track 1 using the 642 nm laser is configured. - In the ‘Channels’ tool define a multi-track experiment using the 642 nm laser for the first channel and the 488 nm laser for the second channel. Adjust laser power to ~10 %, camera gain to 30 V and exposure time of the Andor iXon885 camera to 100 ms to view your sample. Adjust laser power and/or gain if necessary. But use the 'Range Indicator' to avoid overexposure. In the ‘Acquisition Mode’ select the appropriate frame size and set the number of rotations to 5. Adjust frame size further by cropping if needed. If using crop, only crop centrally and do not use panning, otherwise PALM and SIM images will not match later on (Figure 5).
Figure 5. Example of central cropping without panning - Focus on the sample and define the focus as the central slice.
- Using one of the laser lines, define the stack in the ‘Z-stack’ tool using the ‘Center’ method. Use the ‘Optimal’ button to meet Nyquist criteria for the Z sectioning. We recommend setting the slice number to an uneven number to have the focal plane later as the central slice (Figure 6).
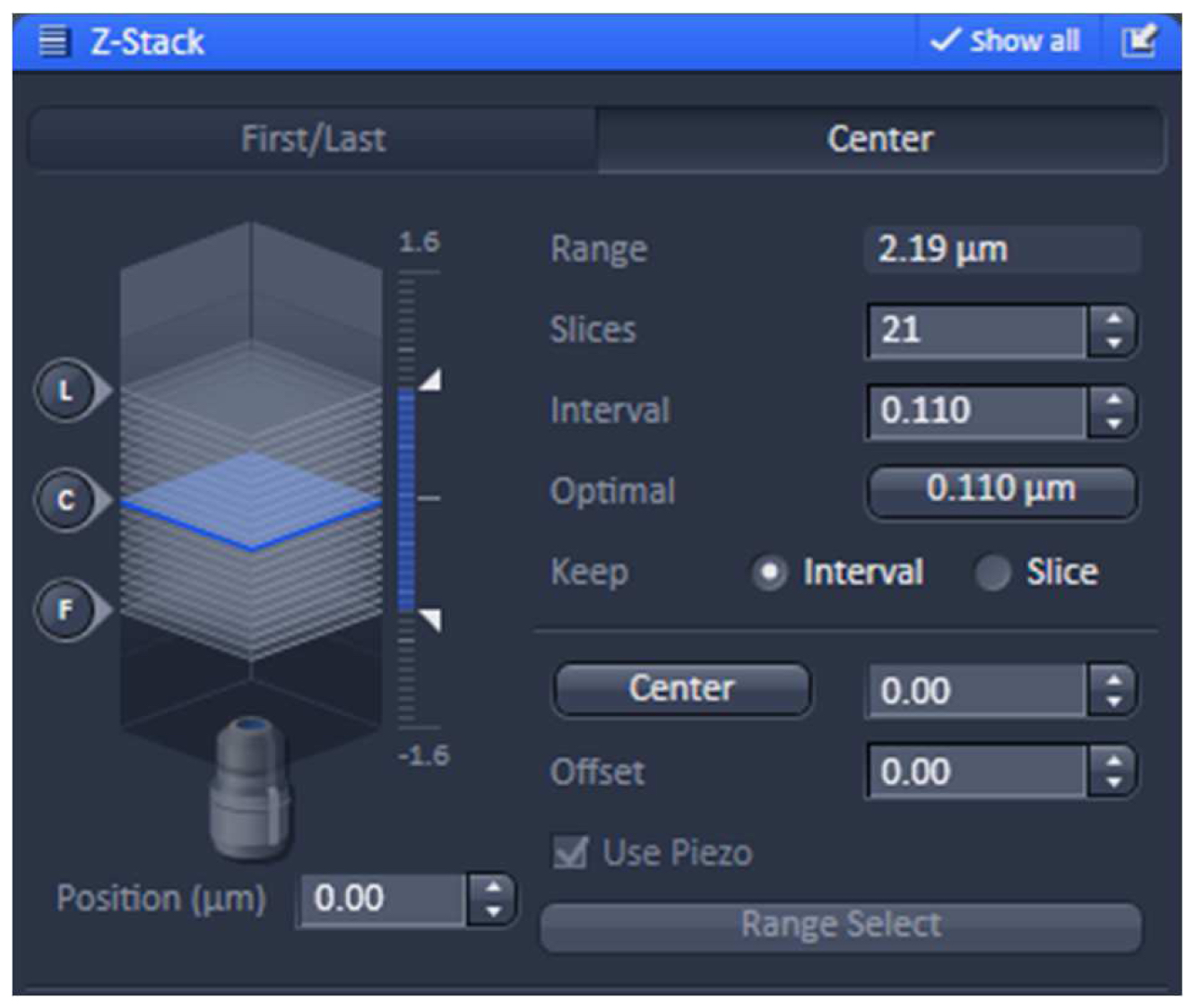
Figure 6. ‘Z-Stack’ tool of ZEN ‘Acquisition’. ‘The ‘Center’ Method is selected. The focus is centered by activating the ‘Center’ button and the optimal slice thickness (0.110 µm) to meet the Nyquist criteria is chosen. - Acquire the image stacks using first the 642 nm line followed by the 488 nm line, which keeps bleaching at a minimum (Figure 7).
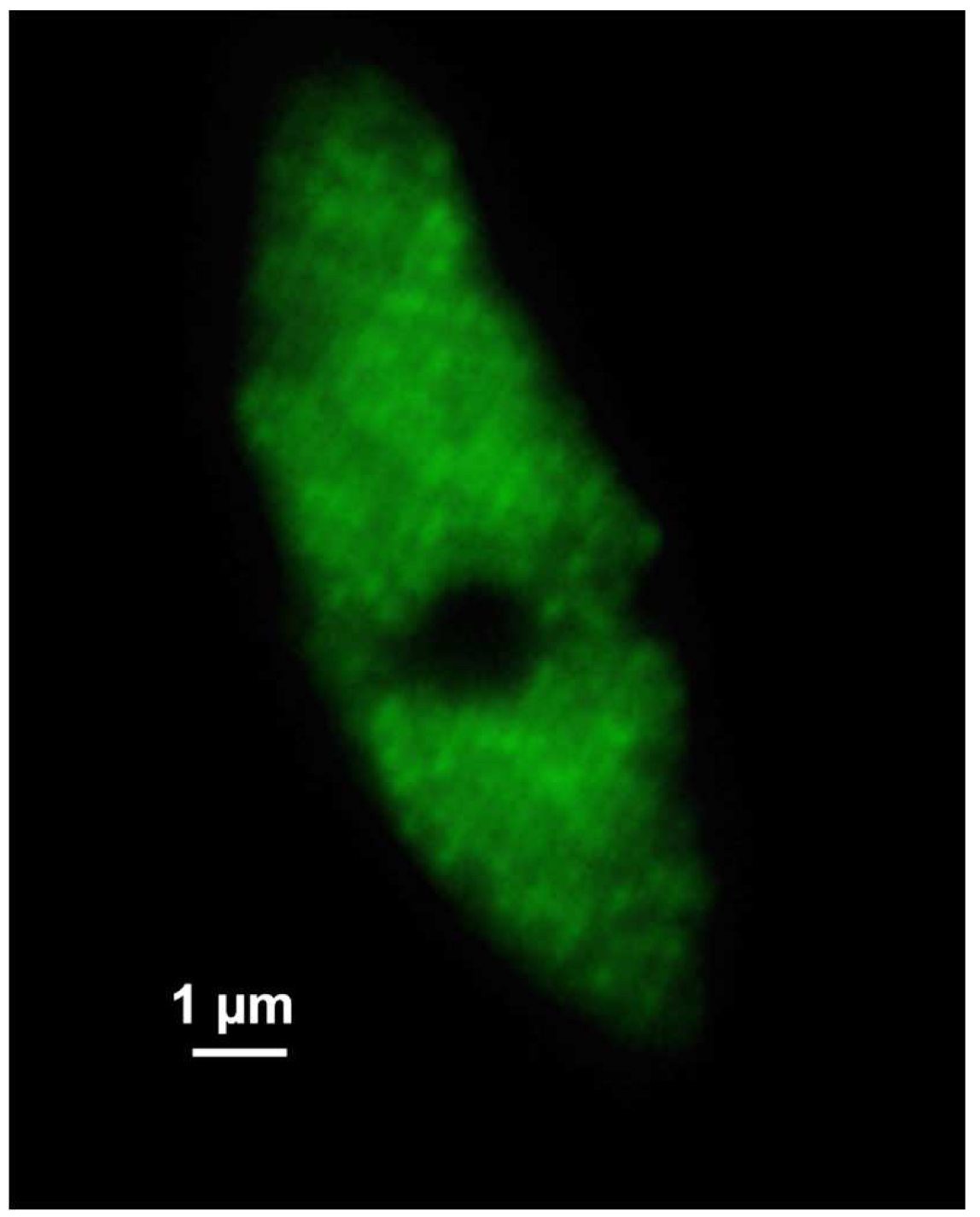
Figure 7. SIM raw data image. Shown is one of the 21 SIM raw slices (5 phases at 5 rotations) from the z-stack of the RNAPIISer2ph sample. The grid projection is visible as stripes on the nucleus. - Save the SIM raw data image stack.
- Switch from tool 'Locate' to the tool 'Acquisition' and select in the ‘Imaging Setup’ tool ‘SIM’ to acquire SIM raw data image stacks (Figure 4).
- Raw data acquisition for 3D PALM
- Select in the ‘Imaging Setup’ tool ‘Laser WF’ to acquire PALM raw data image stacks and activate the ‘TV1: EM CCD Andor PALM’ button (Figure 8).
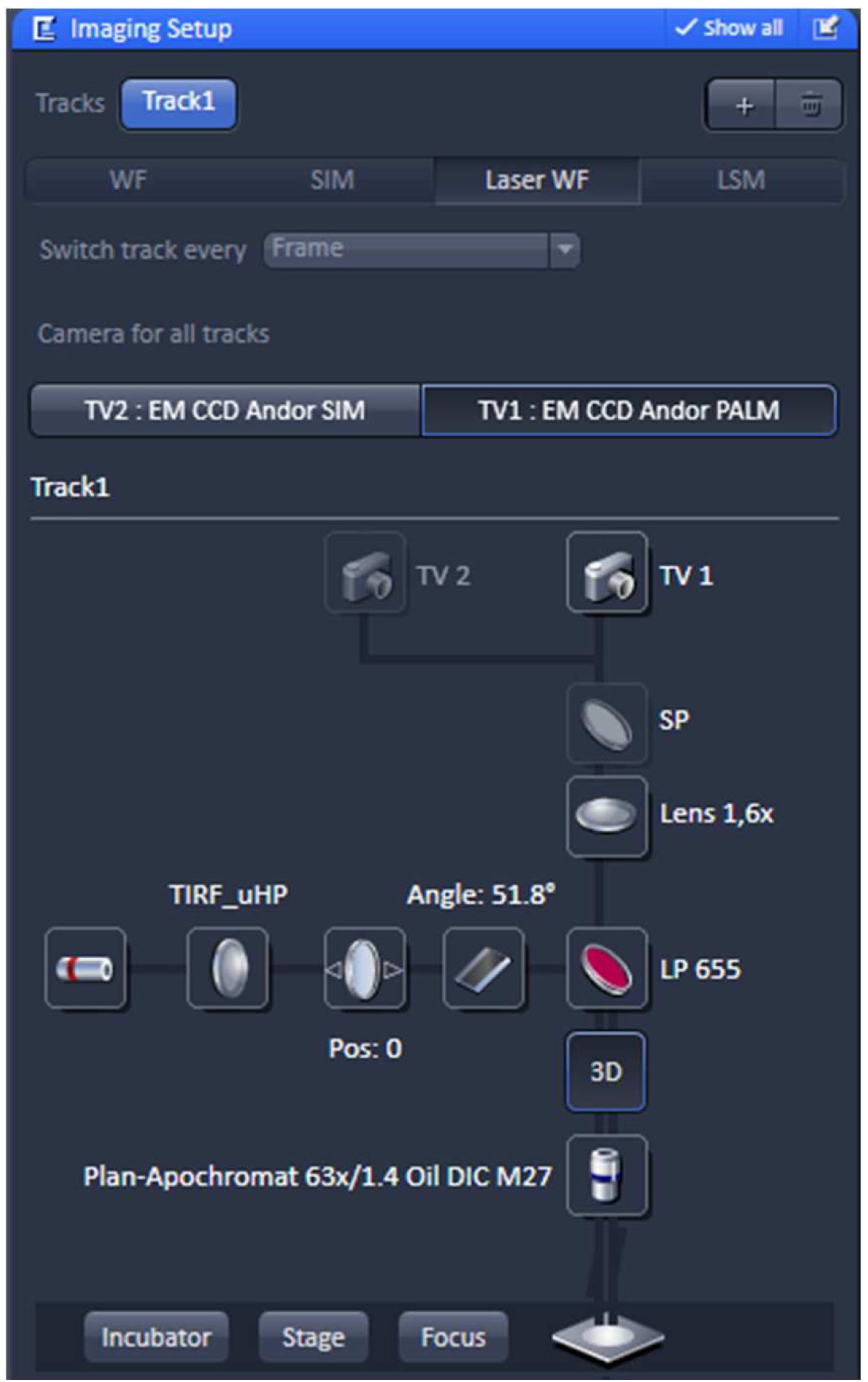
Figure 8. ‘Imaging Setup’ tool of ZEN ‘Acquisition’ for PALM. The experiment is set up for imaging with the 642 nm laser line using the ultra-high power field (uHP). The Andor iXon897 camera is selected by ‘choosing the ‘TV1: EM CCD Andor PALM’ button. - Select the appropriate frame size in the ‘Acquisition Mode’. Since the Andor iXon897 camera to be used for PALM has 512 x 512 active pixels with a pixel size of 16 µm x 16 µm and the Andor iXon885 camera used for SIM has1004 x 1002 active pixels with a pixel size of 8 µm x 8 µm, the frame size in x and y has to be half of that compared to SIM imaging to get finally the same image size. For example, if the SIM image was acquired with 512 x 512 pixels, the PALM image needs to be sampled at 256 x 256 pixels. Do not bin pixels.
- Start with the 642 nm laser line. In the ‘Channel’ tool use a low laser power (up to 5%) and high EMCCD gain (200) to re-focus on the sample using 100 ms integration time. Prominent structures might help in refocusing.
- Switch to the ultrahigh power field (uHP) to increase power density.
- Slide in the 3D PALM slider. Then, activate the ‘Definite Focus’ with continuous stabilization to counteract any z-drift on the touch screen display of the microscope`s stand (Figure 9).
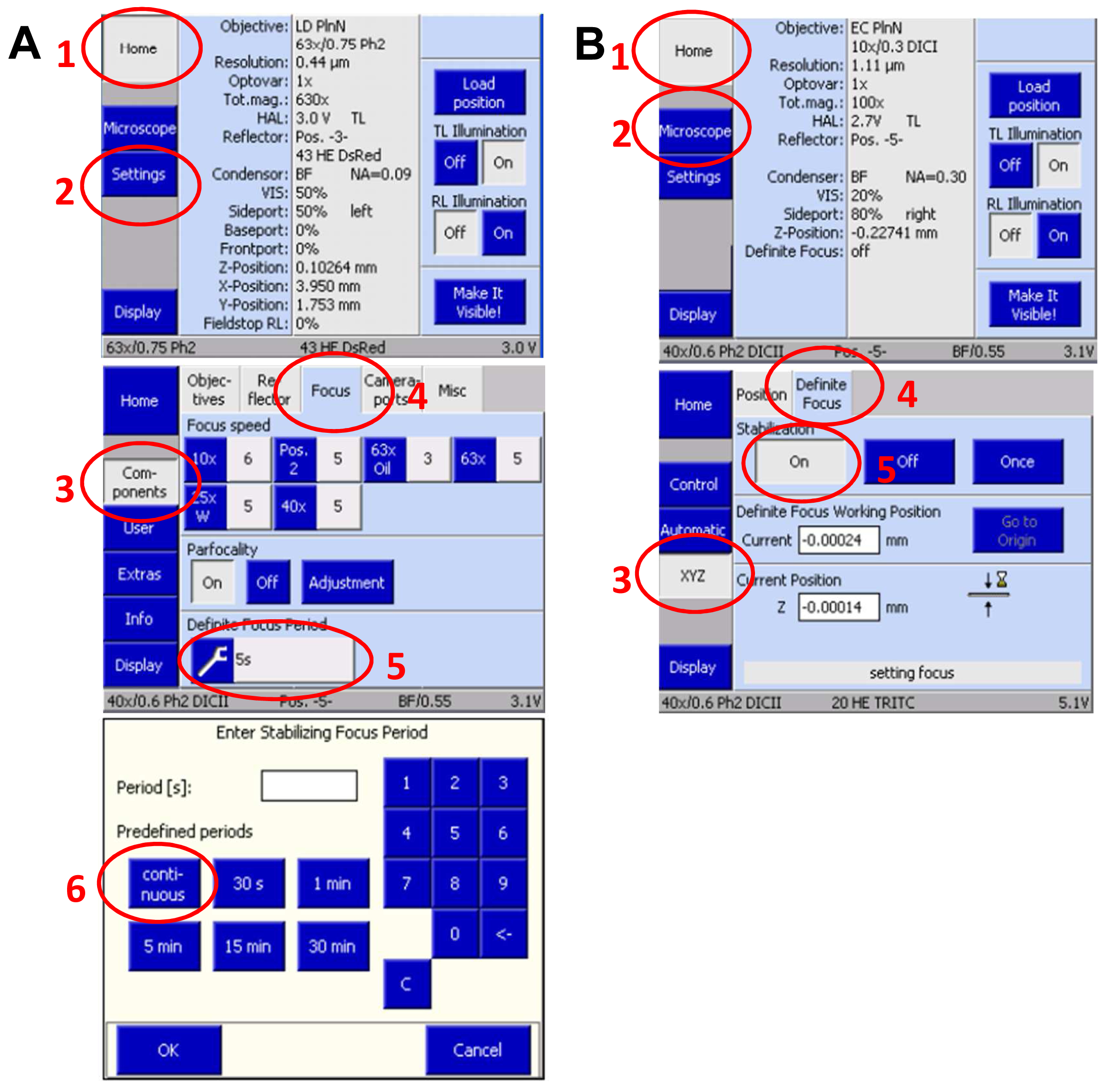
Figure 9. Set up Definite Focus for continuous operation via the touch screen display of the microscope stand. Press ‘Home’ and select ‘Settings’. On the new screen press ‘Components’ and select ‘Focus’. Press the ‘Continuous’ button to set up continuous operation (A). Move in the 3D PALM slider. Then, activate the ‘Definite Focus’ for continuous stabilization to counteract any z-drift by pressing ‘Home’ and then ‘Microscope’ followed by selecting ‘XYC’ and activating the Definite Focus by pressing the ‘ON' button (B). - Define a time series in the ‘Time Series” tool for at least 20,000 frames. This frame number is mostly sufficient to bleach all available molecules. If then blinking does not mainly disappear, increase frame rate correspondingly. Conversely, if all molecules are bleached earlier, you can stop the experiment with fewer frames.
- Decrease the EMCCD gain of the Andor iXon897 camera to 10 V or below. Set exposure time to 30-50 ms. Increase laser power stepwise to 100% to avoid overexposure of the camera.
- Once the molecules are brought continuously to their dark states, increase the EMCCD gain until you reach 200 V without overexposure. You should now see individual molecules blinking. If molecules overlap heavily, wait with these settings until you can distinguish single molecules before recording. If the sample is too dim, increase to 300 V. However, you should be aware that >200 V will increase camera noise as well.
- Start recording the time series. If the sample does not contain fiducials control the temperature. The ELYRA PS.1 has a large incubator box in which approximately 30 °C. will be reached after 2 h. For 2D-PALM experiments in TIRF mode fiducial markers like Tetraspeck beads at a 1:1,000 dilution can be added conveniently to the solution as they settle to the glass surface and can serve as markers for drift correction. For 3D-PALM experiments their use is less helpful because the focus is not near the glass surface. Therefore, the temperature should be as stable as possible to minimize lateral drift. It is recommended to incubate the sample at least 1 h in the Elyra incubator before running the experiment. Save the 642 nm PALM time series (Figure 10).

Figure 10. PALM raw data image. Shown is one of the 20,000 PALM raw images of the RNAPIISer2ph sample. Two events, identified as bi-lobed PRLIM PSF (Phase Ramp Localization Imaging Microscopy Point Spread Function) are encircled. - Repeat steps 3-9 for the 488 nm laser line. Save the 488 nm PALM time series.
- Select in the ‘Imaging Setup’ tool ‘Laser WF’ to acquire PALM raw data image stacks and activate the ‘TV1: EM CCD Andor PALM’ button (Figure 8).
- SIM data processing
- Load the SIM image stacks into the ‘SIM’ Processing tool.
- Select ‘Manual’ processing and within the menu 'Auto Noise Filter', 'Baseline Cut' and 'Theoretical' PSF'. Leave parameters at default values: 'SE Frequency Weighting' at 1.0 and 'Sectioning' at 100/83/83. As output images select 'SR-SIM + Wide-field' to compare the resolution improvement. Otherwise just select SIM image (Figure 11).
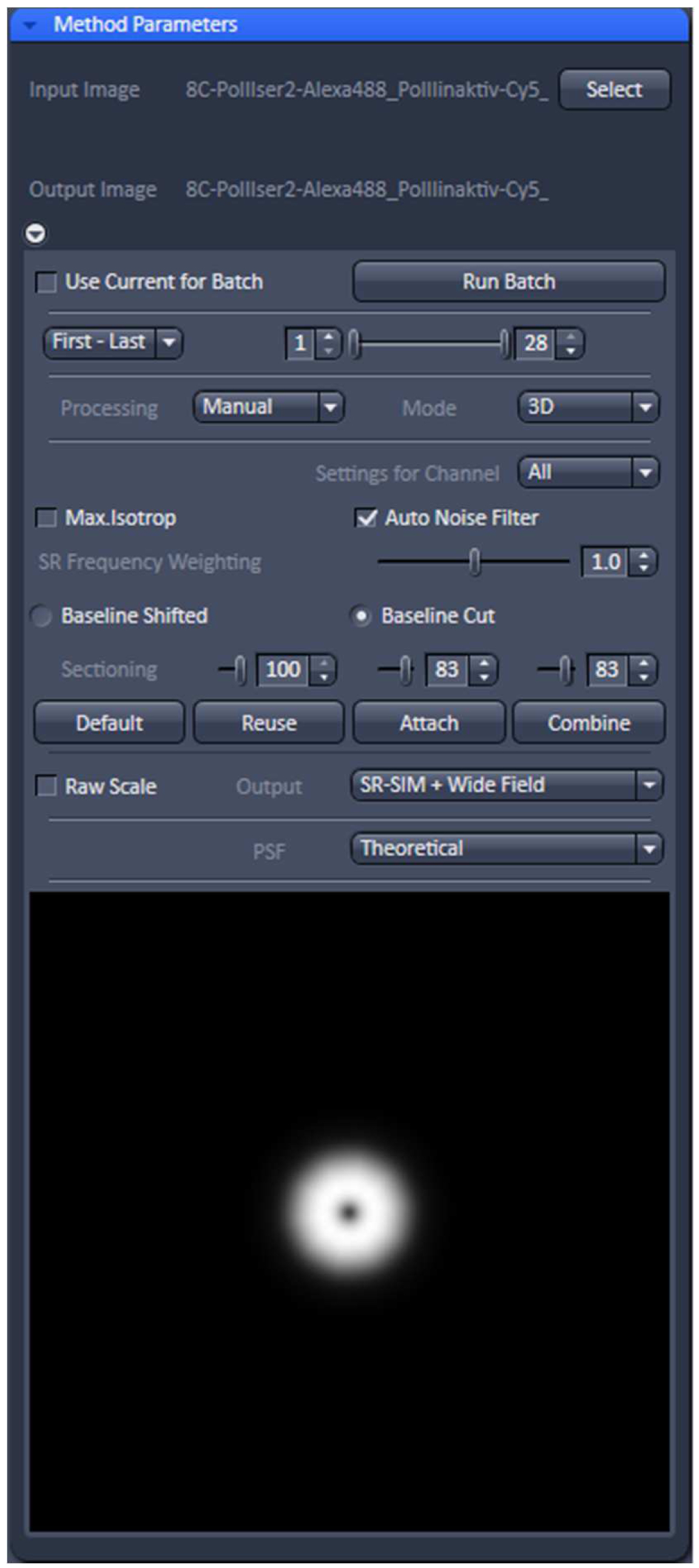
Figure 11. ‘SIM’ processing tool. Display for ‘Manual’ processing with ‘Auto noise filter’ and ‘Baseline Cut’ active and all parameters at default values. The deconvolution will use a theoretical PSF and the output will be a SIM and a Wide-field image stack. - Process the stack for SIM by activating the ‘Apply’ button. Adjust brightness and contrast for each channel and save. Brightness and Contrast are best adjusted by setting the limits to the lower and upper boundaries of the histogram.
- Load the SIM image stacks into the ‘SIM’ Processing tool.
- Determination of the SIM resolution
- If a fine sub-resolution structure is available in the image, you can use it to define the SIM resolution in the sample. Alternatively, TetraSpeck™ Fluorescent Microsphere beads, ideally embedded in the sample, can be used.
- Load the SIM image together with the wide-field image and switch to the ‘Profile’ view tab. Make sure that under ‘Maintain’ in ‘Systems Options’ the ‘Apply brightness and contrast to diagrams and tables’ is selected in the ‘Image display’ tab.
- Identify a fine structure and draw a line across the structure.
- Adjust contrast and brightness to have the baseline at zero and the peak at 40,000 or 60,000 grey values as is convenient. For dim samples the adjustment of the peak will be at lower grey values.
- Export the table of graph to Microsoft Excel and determine the full width at half maximum (FWHM), which gives the resolution (Figure 12).
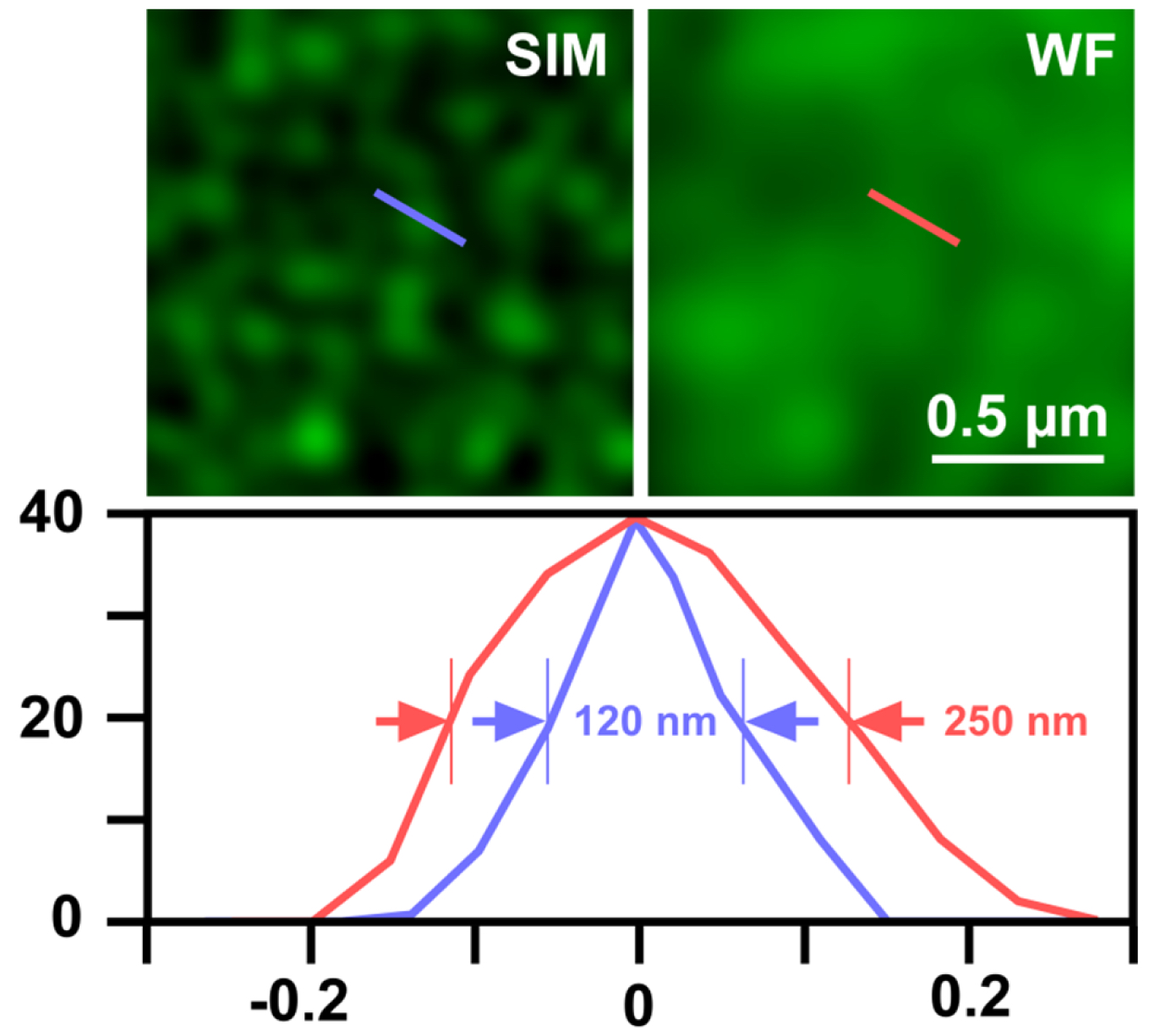
Figure 12. Determination of SIM resolution. Shown is an area of RNAPIISer2ph staining in structured illumination (SIM) and wide-field (WF). The graph below shows a profile through a sub-resolution structure having a size of the expected ~120 nm for SIM compared to ~250 nm in WF, corresponding to a doubling of resolution.
- If a fine sub-resolution structure is available in the image, you can use it to define the SIM resolution in the sample. Alternatively, TetraSpeck™ Fluorescent Microsphere beads, ideally embedded in the sample, can be used.
- Channel Alignment for SIM
- Acquire a z-Stack of 200 nm TetraSpeckTM microspheres spread onto #1.5 coverslips and embedded in a hardening resin of your choice with a step size of 0.11 µm and 41 slices in the relevant colors with SIM and process the data with the ‘SIM’ processing tool. Alternatively, use the 200 nm TetraSpeckTM microspheres slide from Carl Zeiss Microscopy GmbH. This acquired stack will be used to calibrate the color offset of the system. Offsets will be stored in a template that later on can be used to correct the experimental data. Calibrated color offsets are specific for the objective and filters. Therefore, a new calibration is required when objectives and filters are exchanged.
- Load processed SIM data of the beads into the ‘Channel Alignment’ processing tool (Figure 13). Select the ‘Fit’ checkbox in order to align all channels to the first channel. Select either lateral (in x, y and z) or affine (lateral x, y, z & rotational x, y & stretching x, y) correction as needed.
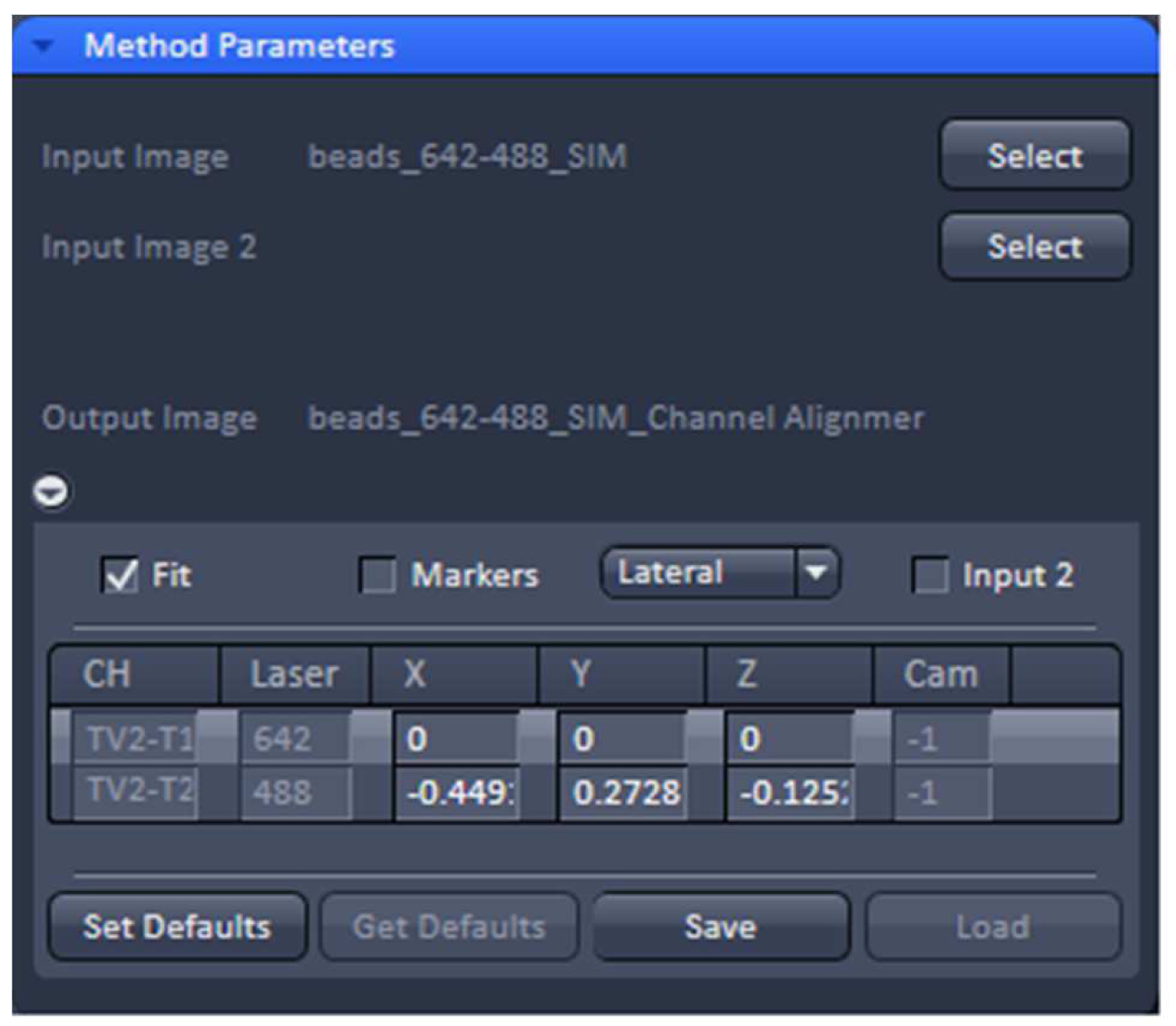
Figure 13. ‘Channel Alignment’ tool of ZEN ‘Processing’. The offset (in pixels) of channel 2 (488 nm) to channel 1 (642 nm) can be stored as a .bin file. - Execute the alignment. The offsets in pixels of all channels in reference to the first channel are displayed. Press ‘Save’ to store the created correction template as a .bin file for later use in order to compensate for any chromatic aberrations and offsets between channels in SIM. Since the ‘Channel Alignment’ tool recognizes the image size as well as zoom and offset position, a template acquired in full frame can be used for cropped sample images.
- Assess the quality of the channel alignment by comparing the multi-color (TetraSpeckTM) beads before and after alignment. Using a highly corrected Zeiss Plan Apochromat 63x oil/1.4 objective chromatic aberrations are low. Nevertheless, slight offsets can be compensated to less than 20 nm deviation between the 488 nm and 642 nm excitations (Figure 14).

Figure 14. TetraSpeckTM microspheres recorded with 488 nm and 642 nm excitation using a Zeiss Plan Apochromat 63x /1.4 Oil objective after SIM processing before and after channel alignment. The slight lateral and even smaller axial offsets were corrected to less than 20 nm. - Load the SIM image stack of your sample into the ‘Channel Alignment’ processing tool as ‘Input’ (Figure 15). Most conveniently the image stack might contain the same channels in the same order as the correction template that is selected for correction. In this way, the determined offsets stored in the template can be applied without the need to assign the correct laser identity (ID) to the channels of the SIM image. If the order is not the same, correct IDs have to be assigned to each channel.
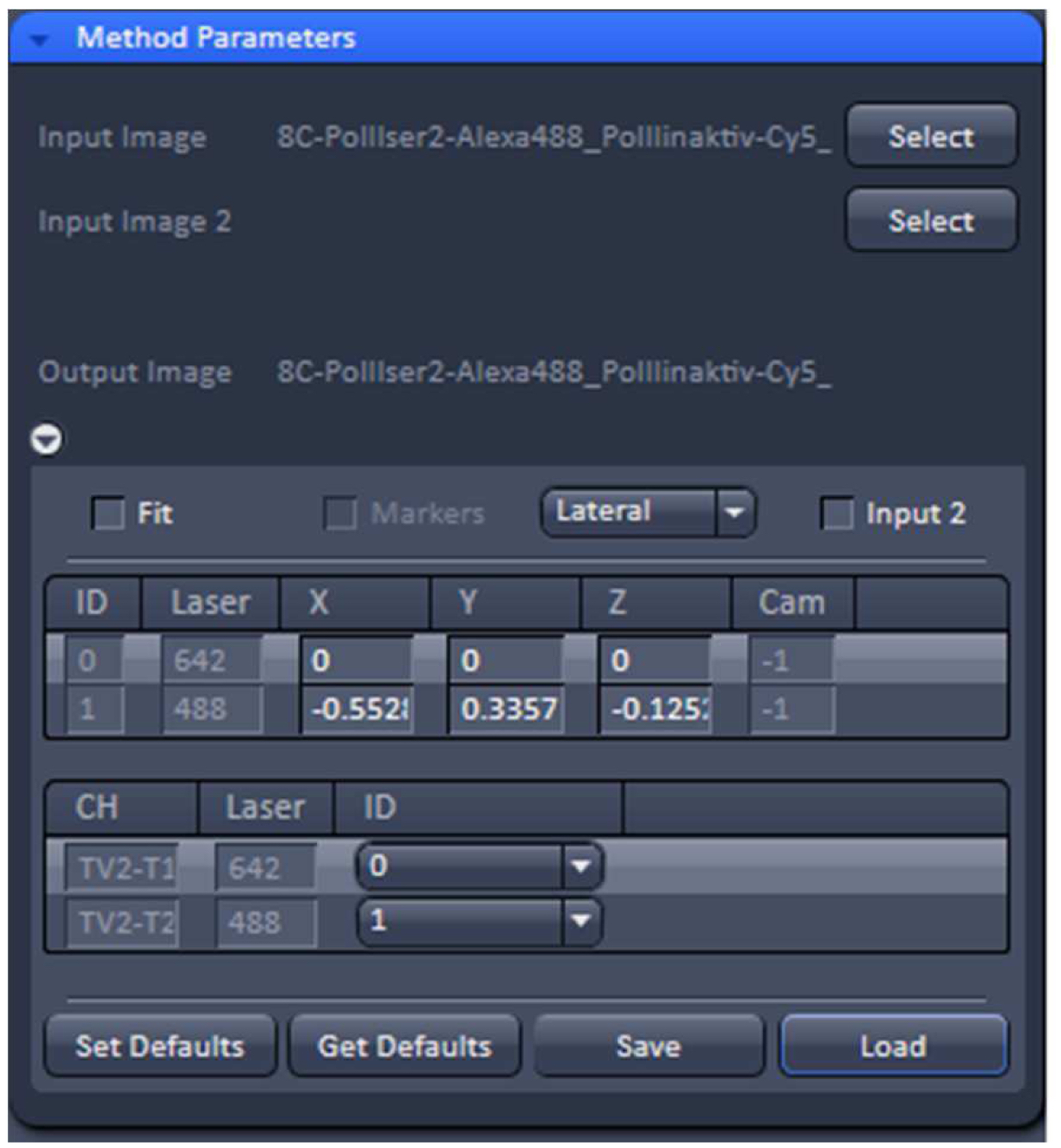
Figure 15. ‘Channel Alignment’ tool of ZEN ‘Processing’. The 2 channel image stacks (excitation with 488 nm and 642 nm) are selected and the correction template with the same lasers in identical order is loaded. In this case the laser IDs of the image stack matches the laser IDs in the template. If this is not the case, the correct laser IDs have to be selected. - Unselect the ‘Fit’ check box and ‘Load’ the correction template (.bin file) to correct the image channels via the values in the correction template. The ‘Fit’ check box should not be selected as otherwise the correction template will not be applied.
- Execute the alignment by activating the button ‘Apply’ (Figure 16). The table will show the offsets between the channels in reference to the first channel. Note, that since the beads used for the correction template are on the coverslip, refractive index mismatches do not play a role. If the sample is also fixed onto the coverslip the obtained correction is reliable and differences in the embedding would not matter. One should be aware, however, that if the sample is fixed on the slide, refractive index mismatches might have an influence. Thus, the correction may be less accurate. In these cases, the beads should be embedded in the same medium as the sample.
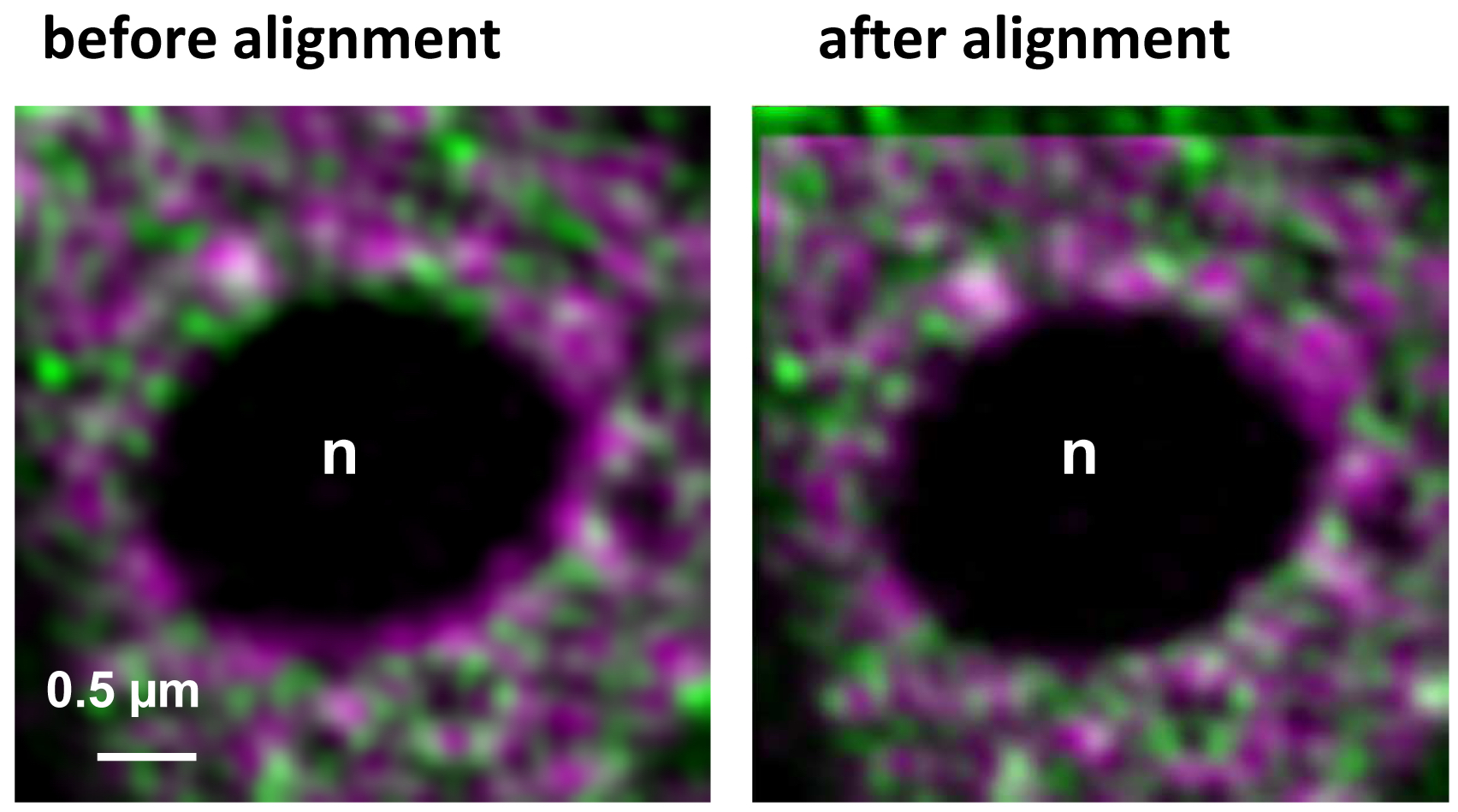
Figure 16. Inactive RNAPII (false colored in magenta) and RNAPIISer2ph (false colored in green) before and after channel alignment using a template. The correction is visible especially at the boundaries of the nucleolus (n) devoid of fluorescence and by the lack of overlap in the left and upper area of the overlay.
- Acquire a z-Stack of 200 nm TetraSpeckTM microspheres spread onto #1.5 coverslips and embedded in a hardening resin of your choice with a step size of 0.11 µm and 41 slices in the relevant colors with SIM and process the data with the ‘SIM’ processing tool. Alternatively, use the 200 nm TetraSpeckTM microspheres slide from Carl Zeiss Microscopy GmbH. This acquired stack will be used to calibrate the color offset of the system. Offsets will be stored in a template that later on can be used to correct the experimental data. Calibrated color offsets are specific for the objective and filters. Therefore, a new calibration is required when objectives and filters are exchanged.
- PSF simulation for 3D PALM data processing
This procedure describes the PRLIM method. For bi-plane, astigmatism and double-helix approaches different procedures apply. The reader has to refer in those cases to specific protocols.- Use an embedded 200 nm TetraSpeckTM microsphere slide at low density as provided by the manufacturer.
- Focus on the beads and use the focal position as the center of the stack. Slide in the 3D-PALM slider. You should see now two equally intense lobes at 45 degrees to each other (Figure 17). If necessary, refocus. Apply the ‘Center’ procedure in the ‘Z-Stack’ tool. Obtain a stack of the beads with the 3D-PALM slider set at 10 nm step size and 401 slices with the respective laser lines. This will ensure sufficient Nyquist sampling.

Figure 17. PRLIM PSF. Appearance of the bi-lobed PSF generated by PRLIM at the focal plane (0 nm), 500 nm below (-500 nm) and above (+500 nm). The shape of the PSF encodes the z-position of the emitter. - Load the stack into the ‘Experimental PSF’ of the ‘Processing tool’ and execute to create an average PSF (Figure 18). When you slide through the stack, only the two lobes should be visible in the focus. If other signals are visible at the edges of the PSF, you must discard it and acquire a new stack at a different position.
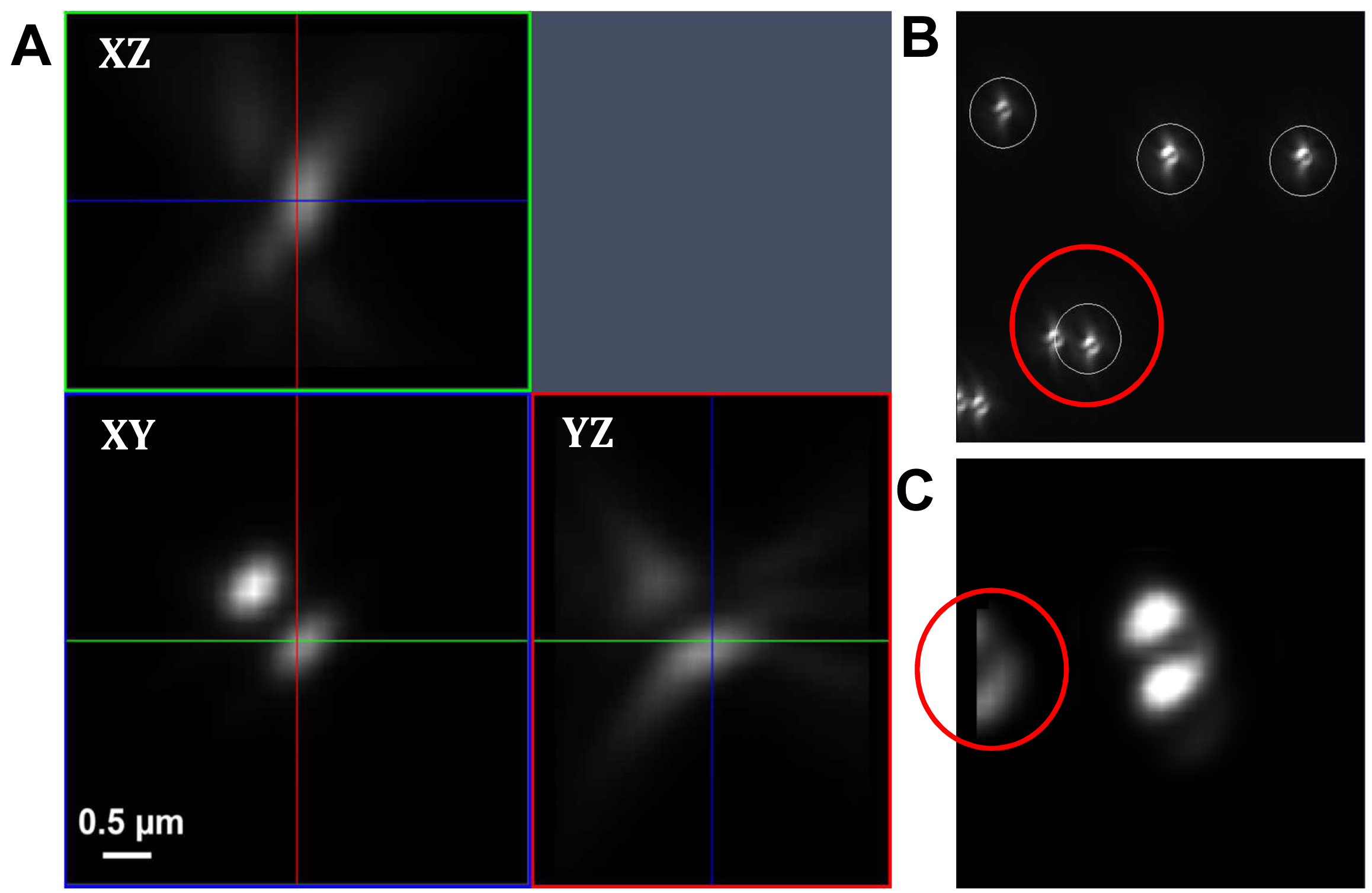
Figure. 18. Experimental PSF. Averaging of many single PSFs in the XY, XZ and YZ view (A). The experimental PSF will be used for the simulations to create the ‘Localization precision table’. In case of two closely lying beads, a part of the second bead might be taken into account (B, red circle) leading to an impaired experimental PSF (C, red circle). Such an Experimental PSF should not be used. Instead, a new stack with less densely lying beads should be acquired. Alternatively, a region excluding such closely lying beads can be cut off from the original image. - Load the average PSF into the ‘Localization Precision’ processing function under the PALM processing menu and execute to obtain a simulated PSF file with z-localization in dependence of the PSF shape.
- Use an embedded 200 nm TetraSpeckTM microsphere slide at low density as provided by the manufacturer.
- PALM data processing, single-molecule counting and distance measurements
This protocol describes the procedure available in the ZEN 2012 software. Software from other suppliers or freely available software might contain identical or similar possibilities. Please follow in these cases the instructions of the supplier.- Load the PALM time series into the ‘PALM’ processing tool (Figure 19).
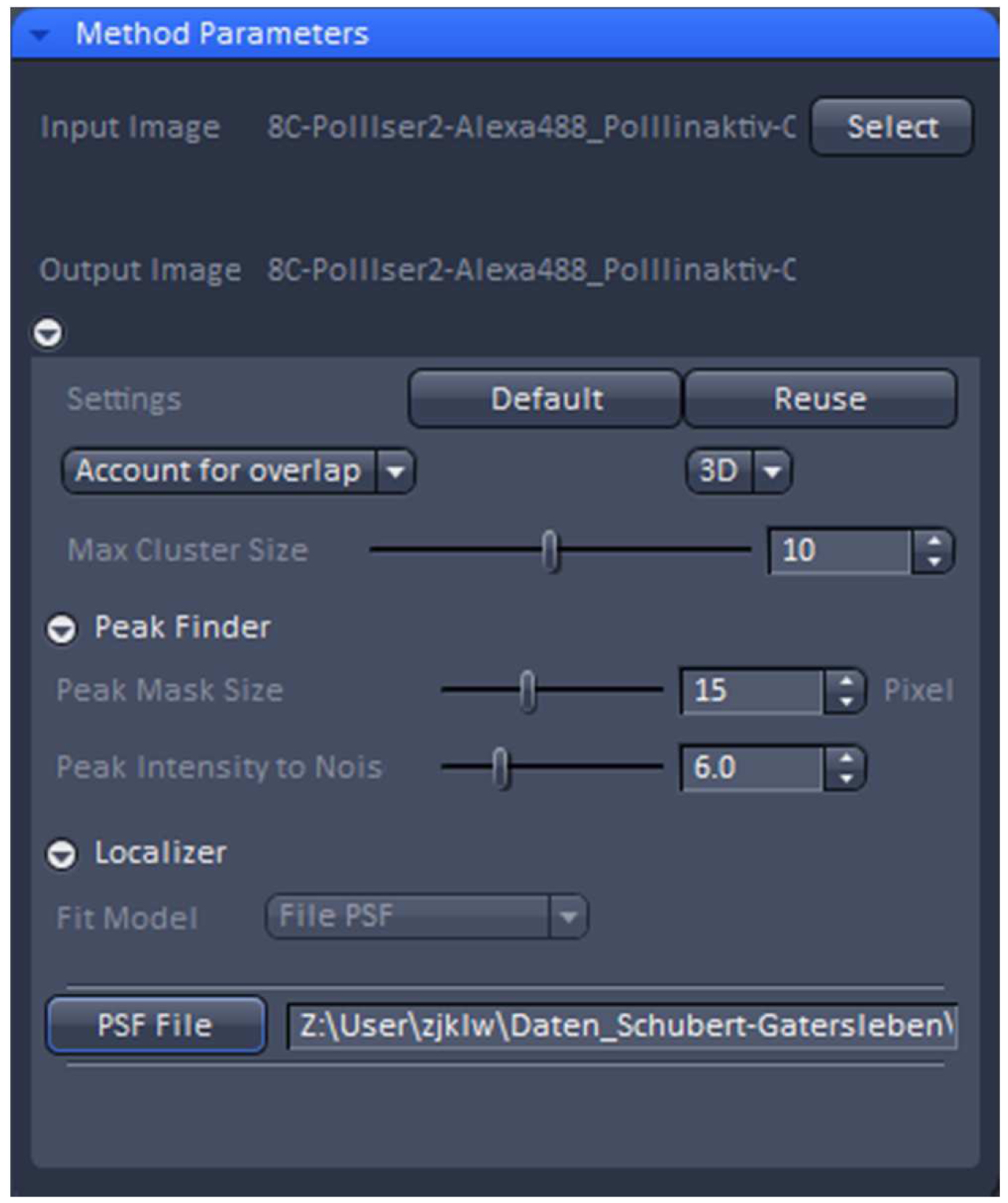
Figure 19. PALM processing tool. A 3D PALM data set was loaded and the parameters set to the displayed values. By activating the ‘PSF File’ button, the PSF with attached 'Localization precision table’ was loaded. - ‘3D’ processing should be automatically selected by the software for a 3D PALM data set. It will show the identified emitters and circles them. Choose ‘Account for Overlap’ for multi-emitter fitting whenever you see intersecting circles, which in 3D-PALM is mostly the case, because its PSF is more spread than a conventional PSF. Use the default value of 6 for ‘Peak Intensity to Noise’ and decrease ‘Peak Mask Size’ from default 19 to 15 to minimize overlapping circles. The 19 is fail-safe, but often the two lobes will fit in a circle of a size of 15. Load the respective simulated PSF file and execute processing.
- With the resulting vector map (PALM image) perform a model-based drift correction in the ‘Automatic’ mode (Figure 20).
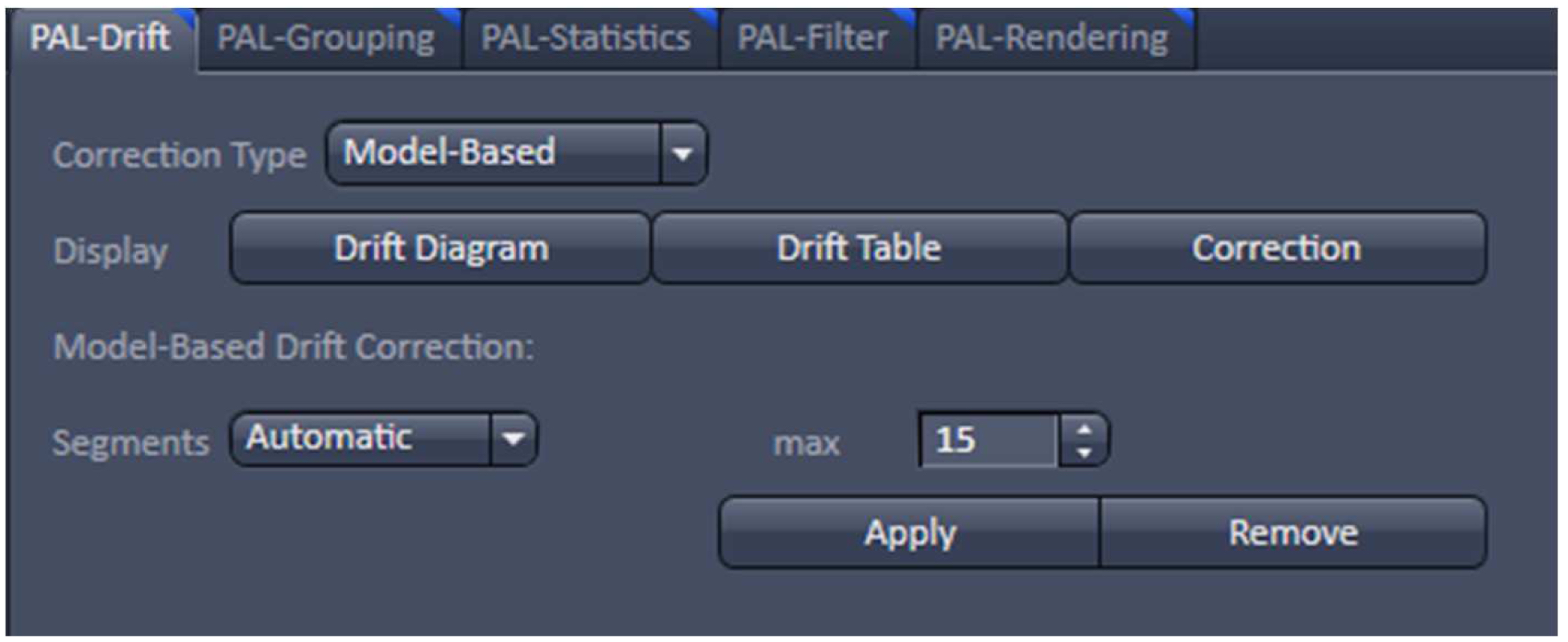
Figure 20. PAL-Drift view tab. Model-based drift correction in 'Automatic' mode selected. - Group the detected events with the ‘Capture Radius’ set to 2 pixels and the 'Max On Time’ and the ‘Off Gap’ sets to 4 and 10 frames, respectively (Figure 21). It is unlikely for most of the dyes that the same molecule is on for more than 4 frames or off for more than 10 consecutive frames under the applied imaging conditions. Also, if two events are not within a distance of two pixels, it is unlikely that the two events are caused by the same molecule.
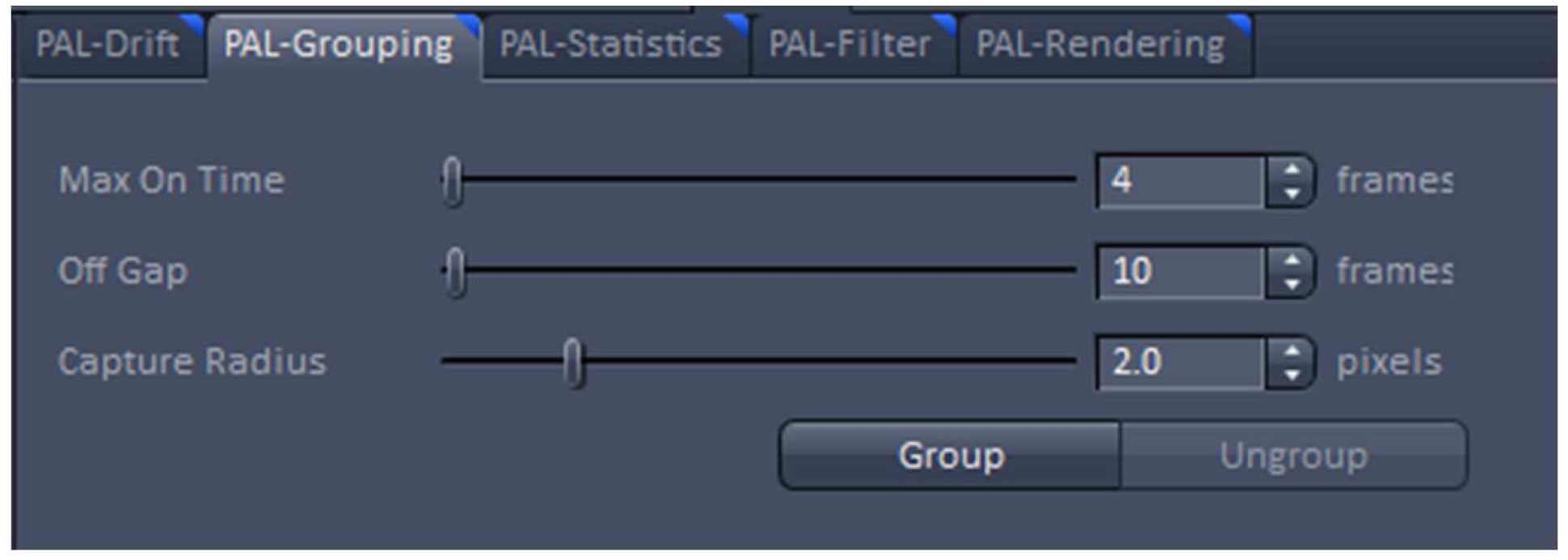
Figure 21. PAL-Grouping view tab. Definition of filters that determine, which events are considered to be derived from the same emitter. - Render the image at 10 nm x, y pixel size and at 40 nm in z to obtain a Nyquist sampling with a resolution of 20 nm laterally and 80 nm axially, corresponding to the maximal resolution which can be obtained by 3D-PALM (Figure 22). If needed, you can filter the data. For example, if you want to display data which show a localization precision between 10 nm and 40 nm laterally and between 10 nm and 80 nm axially, set the boundaries accordingly for the 'Localization precision' and the 'Localization precision z' in the PAL-Filter tool (Figure 23). If there are enough well localized molecules, this yields a higher resolved image. Store the PALM vector map.
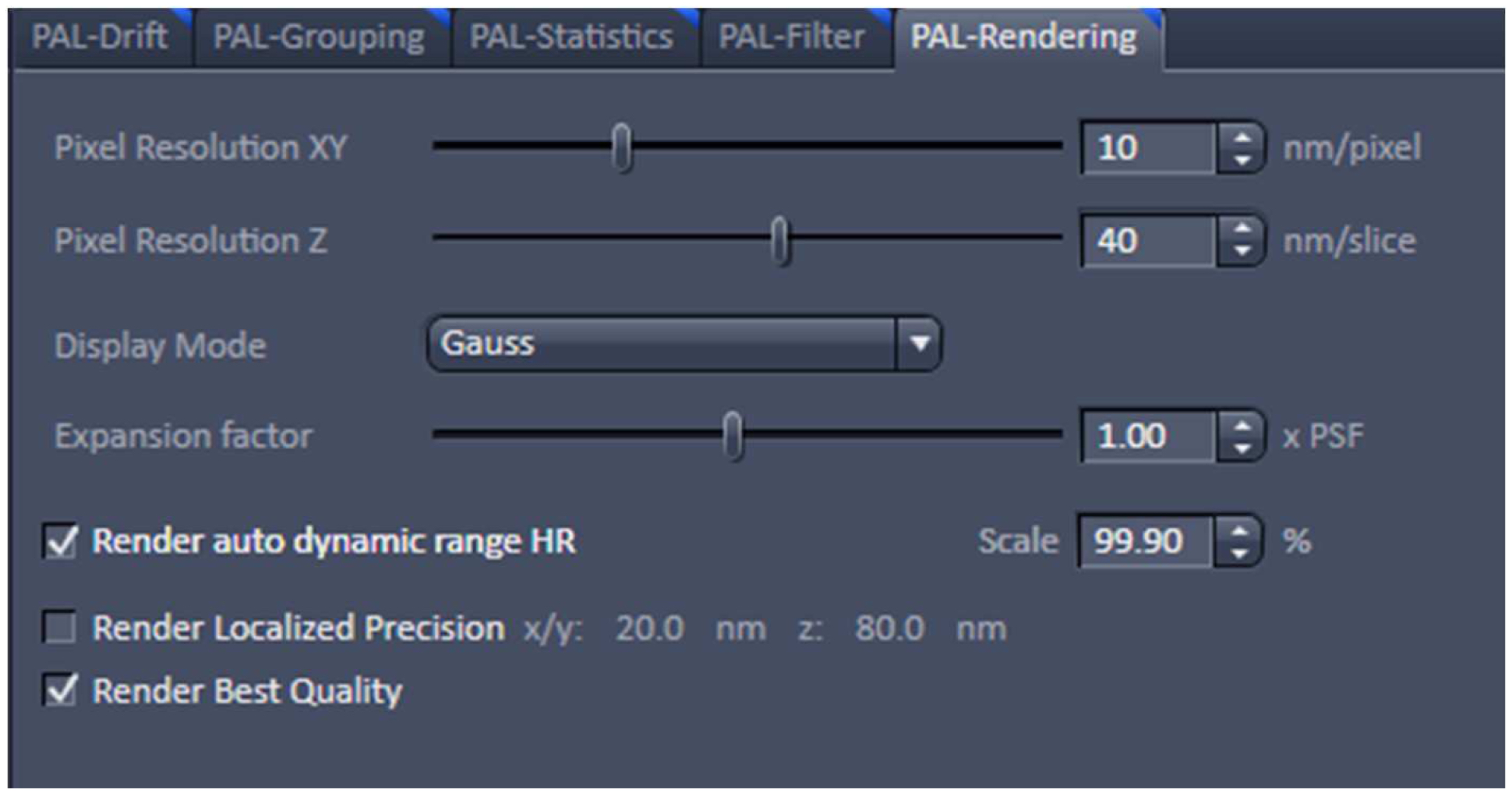
Figure 22. PAL-Rendering view tab. Selection of `Pixel Resolution’ and ‘Display Mode.’ - Define a region of interest using the ‘Graphics’ tool view and select the ‘Use Region(s)’ in the 'PAL-Filter’ view tab to define x, and y dimensions. Set additionally the ‘PositionZ’ range (Figure 23).
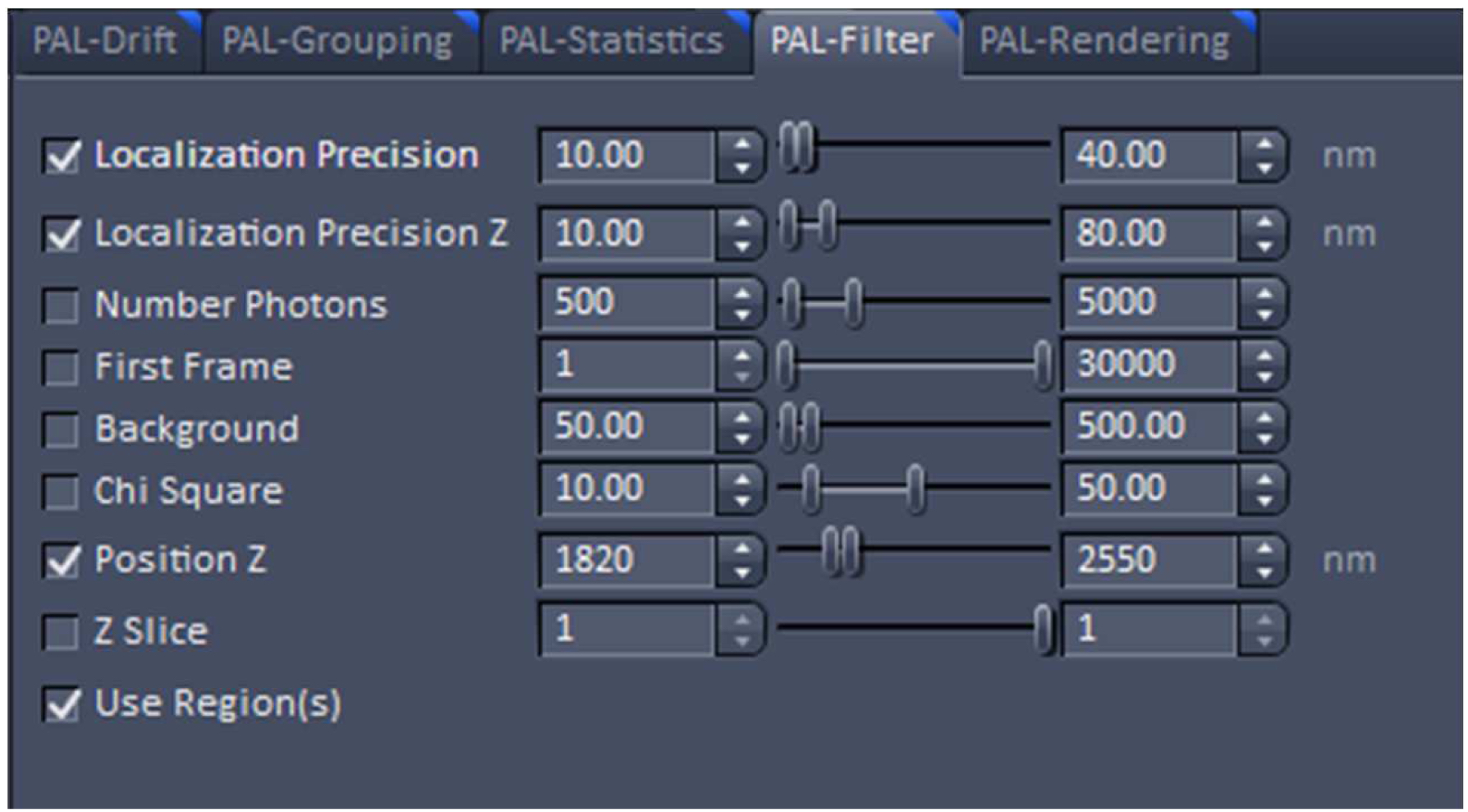
Figure 23. PAL-Filter tab view. The active check-boxes will apply the displayed settings to the image. - Activate ‘Table’ in the ‘PAL-Statistics’ view tab (Figure 24) and scroll down the table to the last entry showing the number of the detected molecules (Figure 25). Please note, that grouping is advisable to avoid counting molecules twice and more. The histogram (Figure 26) can be assessed to estimate the distribution of the parameter values, it can help to set the filters and to check the quality of the data. For example, the histogram for the localization precision will inform about the achievable resolution in the image. In the example shown in Figure 26, the peak is around 40 nm, which is about 10-fold better than the diffraction limited resolution. Furthermore, the histogram indicates that there are few molecules of very high localization precision values, which can be filtered out by setting the respective limits in the PAL-Filter tab view (Figure 23).
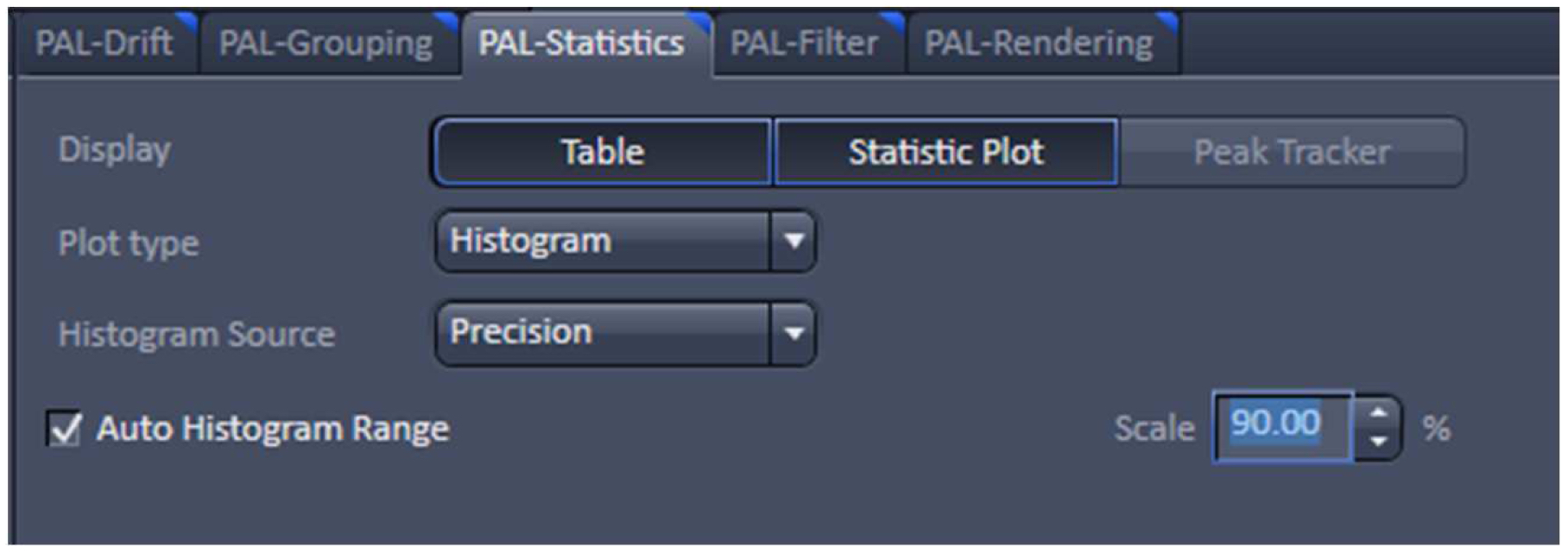
Figure 24. PAL-Statistics view tab. Pressing the ‘Table’ and ‘Statistic Plot’ tabs will display the molecule table with parameters and histograms / scatter plots of the selected parameters.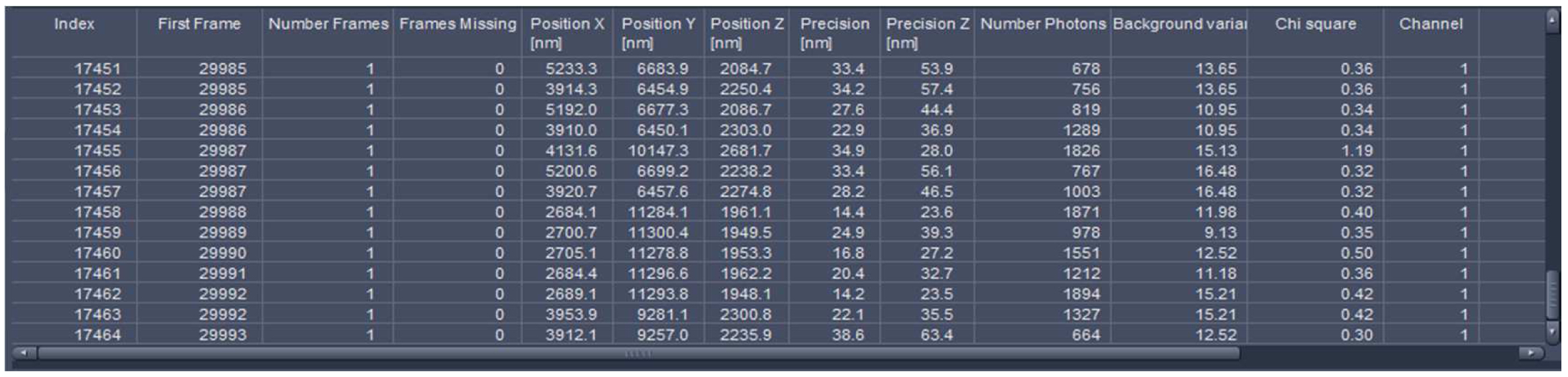
Figure 25. Statistics table. The table lists all events with the respective parameters. The last index indicates the total number of detected molecules.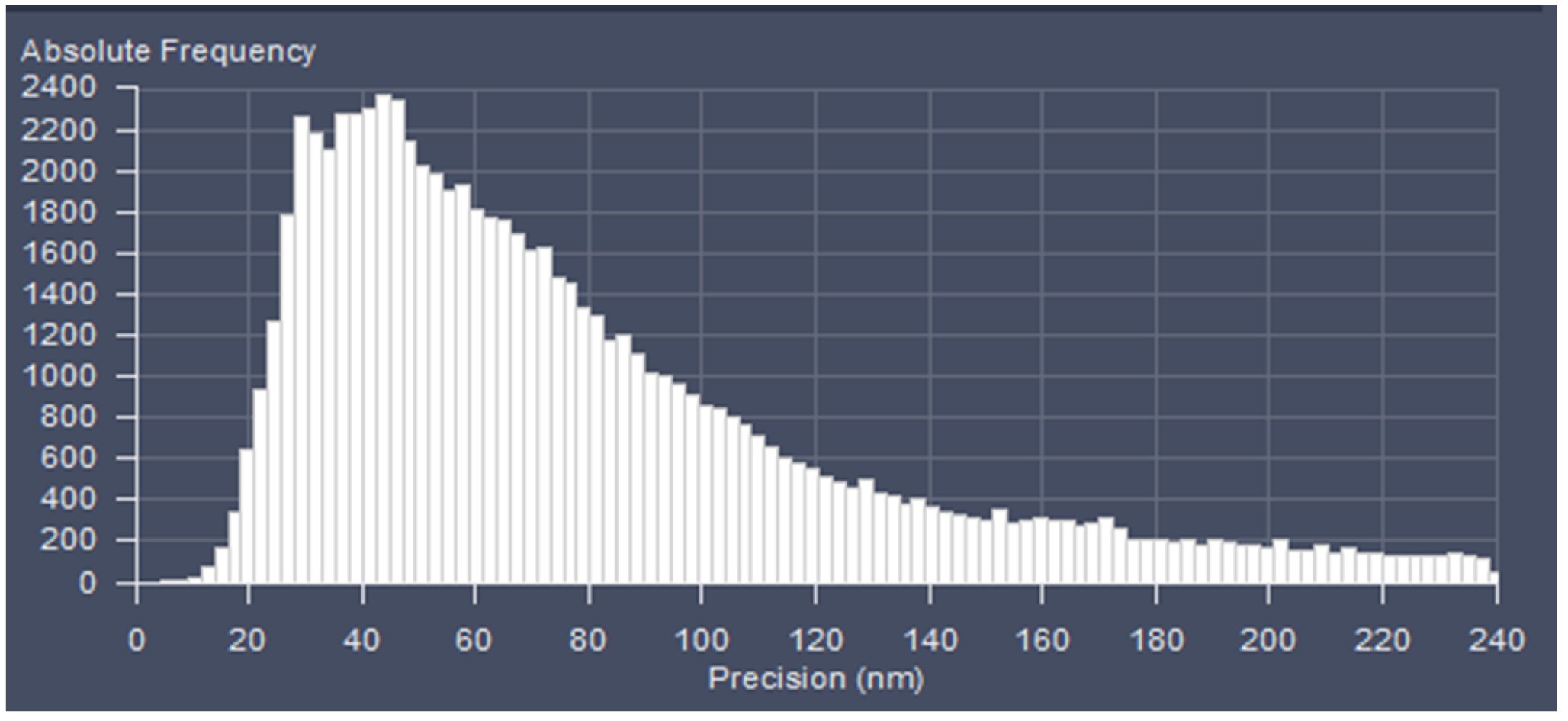
Figure 26. Histogram. The histogram shows the value distribution of the selected parameter. In this example the localization precision is displayed. - To measure distances, display the localized molecules as centroid or Gaus-render with 1 pixel in the ‘PAL-render’ view tab. Use the ‘Graphics’ line tool to measure distances between two localized molecules (Figure 27).

Figure 27. Distance measurement of molecules. PALM vector map with Gaus + cross displayed. Distances are measured by drawing a line between two crosses using the ‘Graphics’ tool. Then, the length is displayed. - If clustering is observed (best seen with a Rainbow look up table when molecules are Gaus rendered), draw a circle or ellipsoid using the graphics tool to approximate the outlines of the cluster. Please ensure that the outline is approximated as best as possible. To our experience the measurement error stays within ±10%. Determine the mass of center by the intersection of two perpendicular diameters (circle) or the two axes (ellipsoid). Use the centers to determine the distance between clusters (Figure 28).
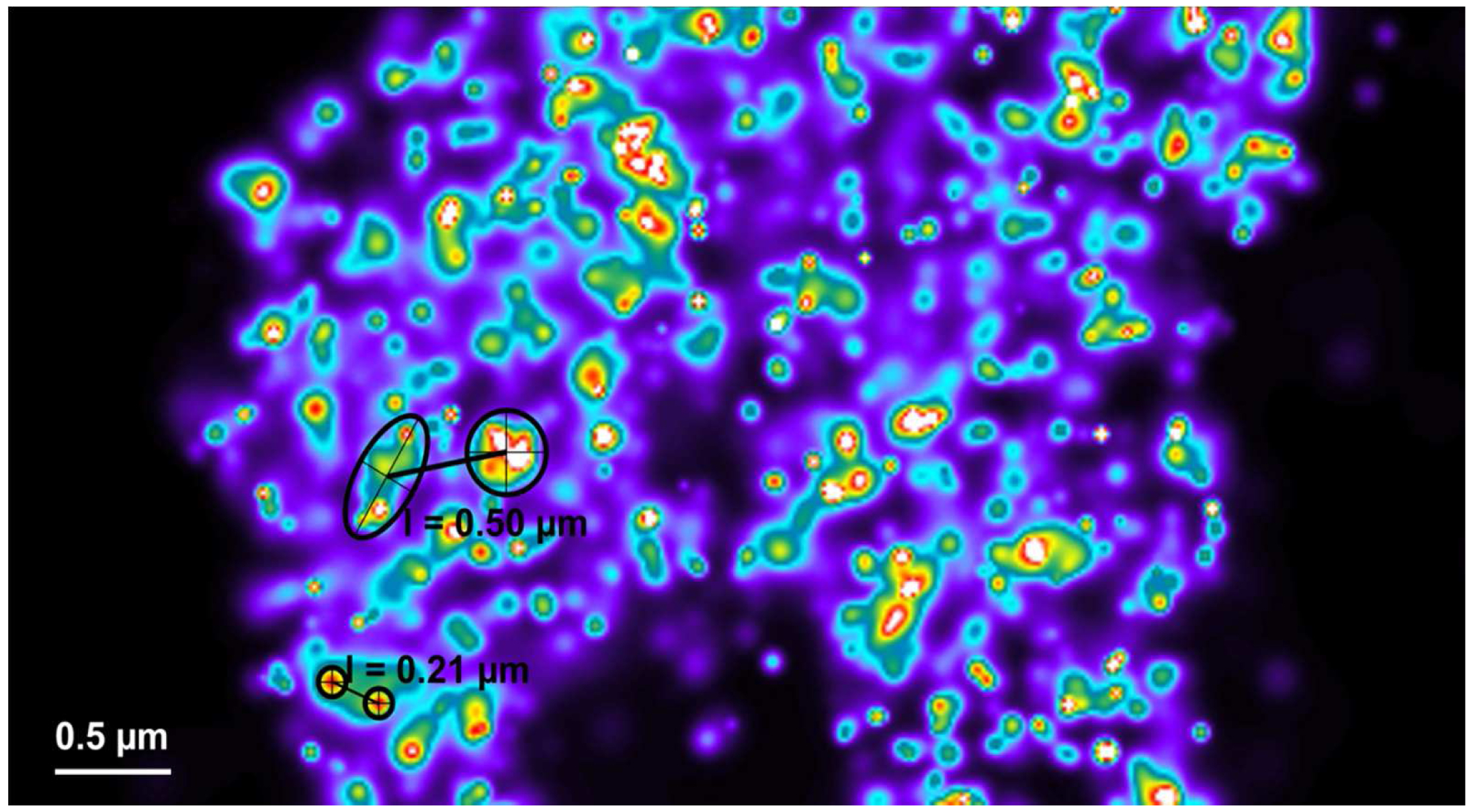
Figure 28. Distance measurements between clusters. Clusters can be most conveniently identified by a Rainbow look up table. They can be approximated by circles or ellipses as shown (in black) for small (small circles) or big (large circle and ellipse) clusters. Their mass center can be determined by the intersection of two diameters (circles) or axes (ellipse). Connecting the centers by a line from the ‘Graphics’ tool will display the length of their distance.
- Load the PALM time series into the ‘PALM’ processing tool (Figure 19).
- Channel Alignment for PALM and co-localization of molecules
- Load the PALM vector maps (tables) into the ‘Convert to Image’ processing tool of the PALM processing functions and execute the conversion of the vector map into a ‘czi’ image format. 'Convert to Image' preserves the voxel resolution as defined in the PALM data set. Please note that the ‘Channel Alignment’ processing function will work on PALM vector maps only if fiducial markers are present.
- To merge the two PALM channels use either the ‘Copy’Channels’ or the ‘Channel Alignment’ processing tool. In the latter case, load the first converted PALM image as ‘Input’ and the second one as ‘Input 2’. If the 'Fit' check box is unselected and the 'Input 2' check box active, the two channels will be just copied together without a fit application after activating 'Apply' (Figure 29).
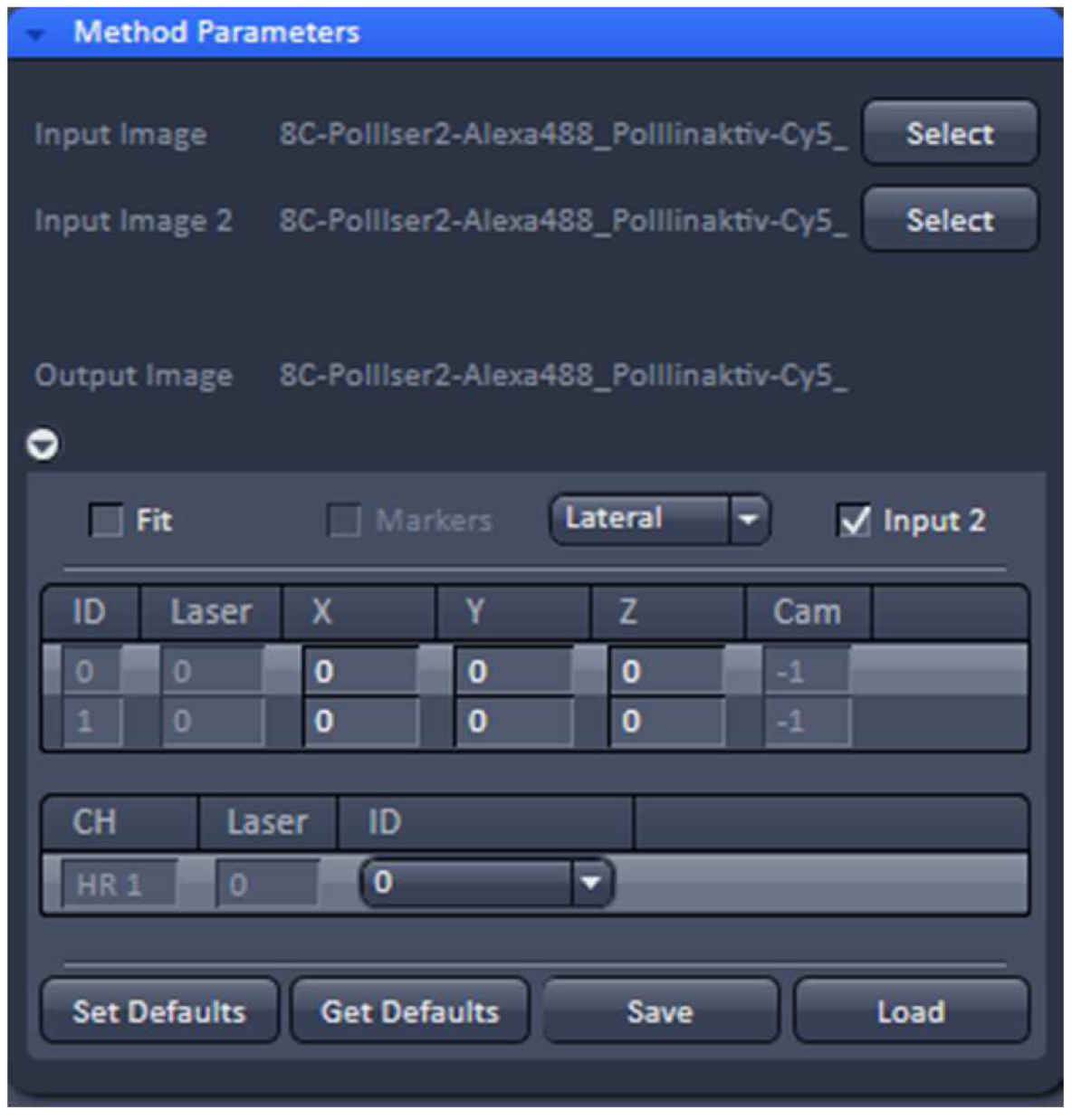
Figure 29. ‘Channel Alignment’ tool of ZEN ‘Processing’. Two channels are loaded as ‘Input Image’ and ‘Input Image 2’. If the ‘Fit’ box is not selected and the ‘Input 2 box’ selected, then the two channels will be copied together into a merged two-channel image.- Load the merged channels into the ‘Channel Alignment' tool. Select the 'Fit’ button to align the second channel to the first channel. Please note that drift between the two consecutive PALM images is also interpreted as a color offset. When no fiducials are used it is essential that drift is kept at a minimum to avoid any contribution to color offset. Z-drift was compensated with the ‘Definite Focus’, whereas lateral drift was largely minimized by pre-incubation of the sample for at least 1 h in the incubator and keeping the temperature in the incubator constant. Thus, the lateral drift was less than 50 nm over a period of 25 min (Figure 30). If significant structures, like nucleoli devoid of fluorescence, are present, they can be used for channel alignment by drawing a region of interest around them.
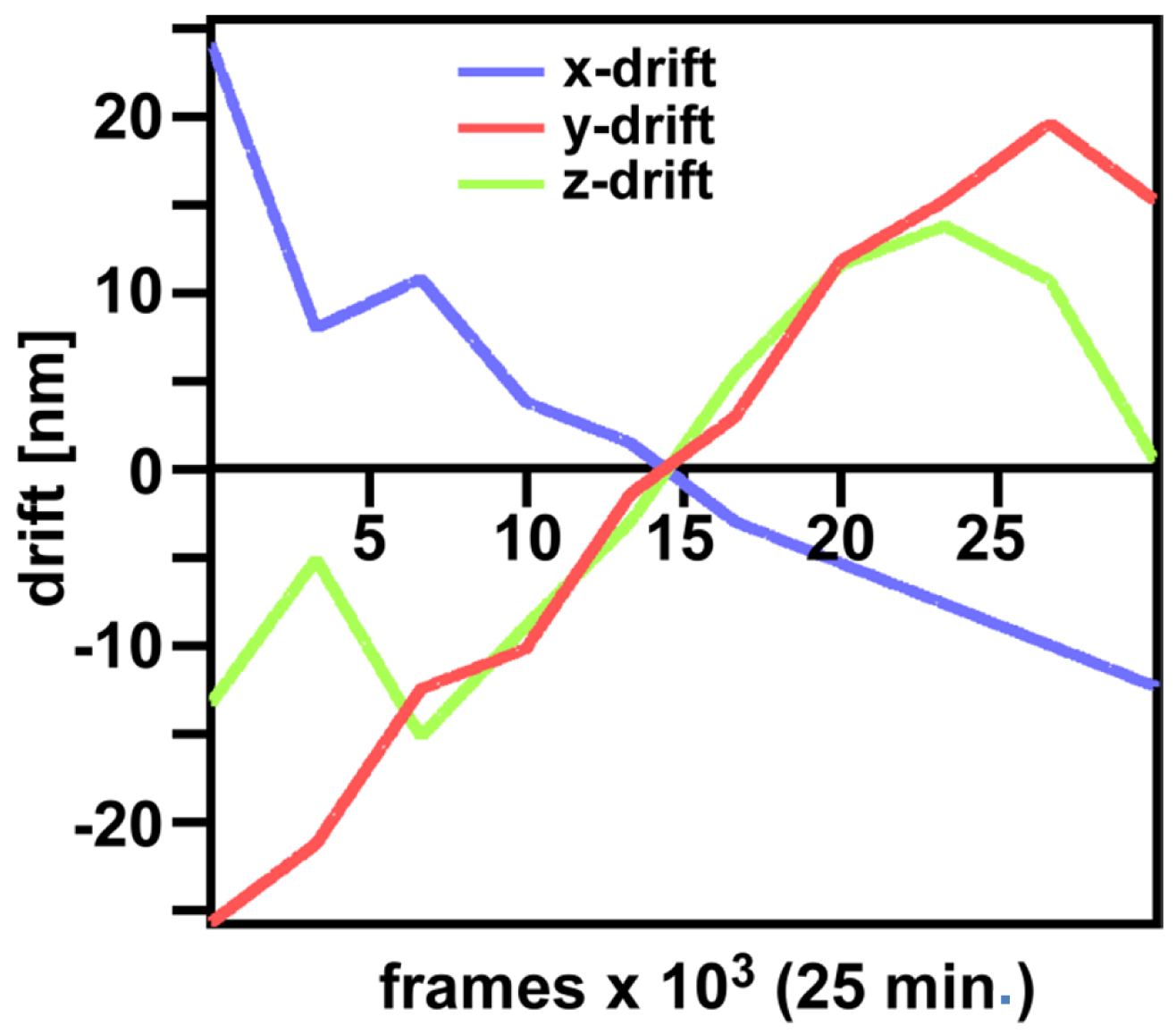
Figure 30. Drifts obtained with ‘Definite Focus’ and pre-incubation of the samples. Frame time was 50 ms.- Fine adjust color correction by using 'Channel Shift' of the ‘Adjust’ processing tool under ‘visual inspection’, if significant structures can be used as landmarks for the alignment (Figure 31). Alternatively, if drift was minimal (≤100 nm), the 'Channel Alignment' tool with the correction template loaded can be used as described for SIM images.
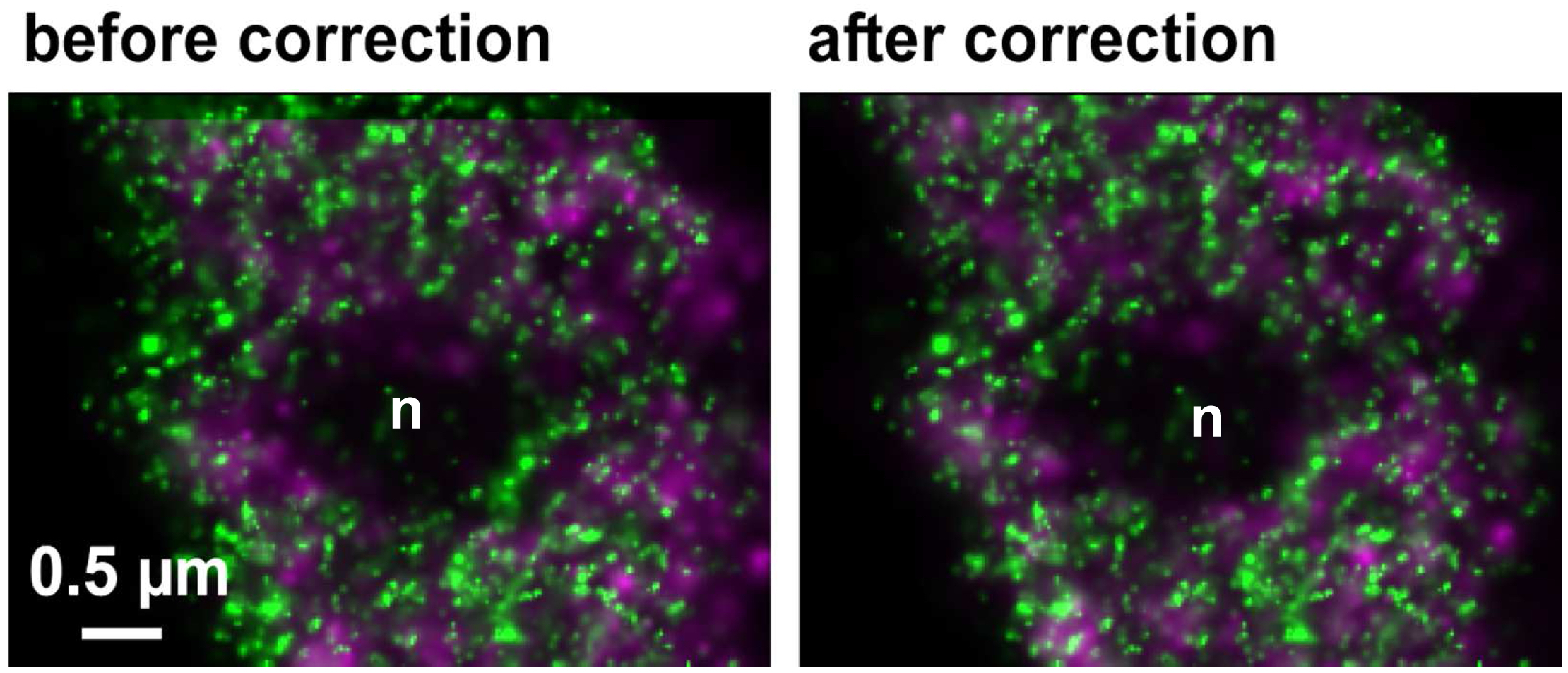
Figure 31. Inactive RNAPII (false colored in magenta) and RNAPIISer2ph (false colored in green) before and after drift correction and channel alignment followed by visual inspection based on structural landmarks (e. g. nucleolus = n)- Pseudo-color channels, conveniently in green and magenta for co-localization. Look for co-localization (appears in yellow). But any colors with a distinct mixed color will do. For quantitation use the 'Coloc' tool of ZEN.
- Load the PALM vector maps (tables) into the ‘Convert to Image’ processing tool of the PALM processing functions and execute the conversion of the vector map into a ‘czi’ image format. 'Convert to Image' preserves the voxel resolution as defined in the PALM data set. Please note that the ‘Channel Alignment’ processing function will work on PALM vector maps only if fiducial markers are present.
- Alignment of SIM and PALM images
- Set the slice thickness of the PAL vector map identical to the slice thickness of the SIM image in the Render view tab (Figure 22).
- Convert the PALM vector map into a 'czi' format image using the ‘Convert to Image’ PALM processing tool.
- Copy the SIM and converted PALM image together using the ‘Copy Channels’ tool and load into the ‘Channel Alignment’ tool (Figure 32). Alternatively load the SIM and converted PALM images as ‘Input’ and ‘Input 2’ into the ‘Channel Alignment tool’.
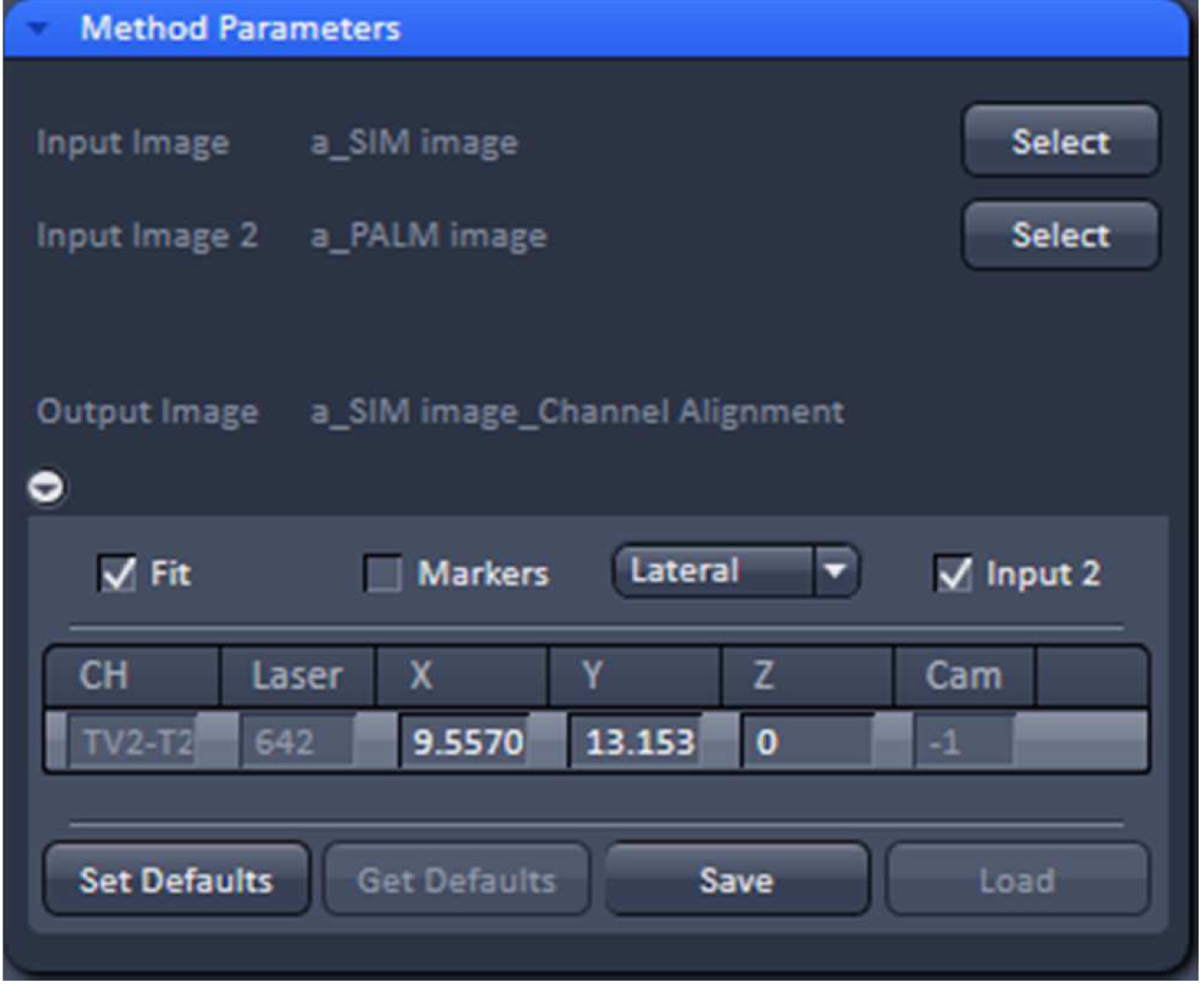
Figure 32. 'Channel Alignment' tool of ZEN ‘Processing’ to align SIM and PALM images representing the same structures - In the latter case, when the SIM and PALM channels represent the same structures, the alignment tool can be used to fit the channels together if the 'Fit' and 'Input 2' boxes are selected. For the ‘Copy channel’ image only the ‘Fit’ box has to be selected. If prominent structures are present, define a region of interest for the alignment. Execute the alignment (Figure 33).
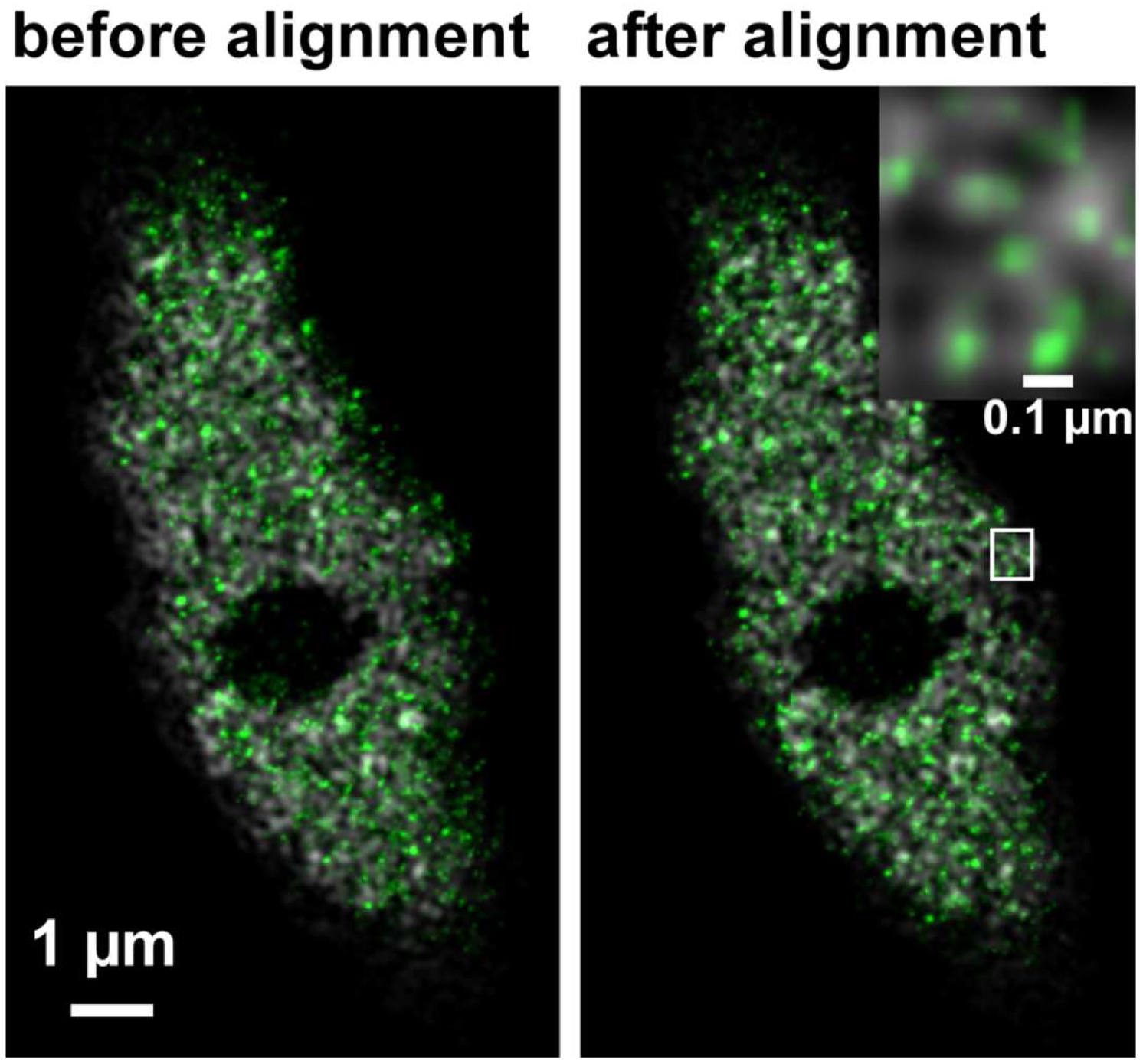
Figure 33. Overlay of the PALM (color coded in green) on SIM image (color coded in grey) of RNAPIISer2ph before and after channel alignment. The inset on the right upper corner is a magnified view of the boxed area and displays the localization of RNAPIISer2ph molecules Gaus rendered within the reticulate structures revealed by SIM. - If necessary, employ prominent structures for fine adjustment by visual inspection using the 'Channel Shift' of the ‘Adjust’ processing tool group.
- Set the slice thickness of the PAL vector map identical to the slice thickness of the SIM image in the Render view tab (Figure 22).
Notes
- The alignment of PALM and SIM images in 3D needs careful adjustment of the instrument for data acquisition. You have to ensure that the image sizes are the same. If you are using the Andor iXon 885 camera with its 8 µm pixel sized chip for SIM and the Andor iXon 897 camera with its 16 µm pixel for PALM, the frame size for PALM has to be half (8 µm/16 µm) compared to SIM. Thus, if the SIM image was recorded with a frame size of 512 x 512, the PALM image must be taken with a frame size of 256 x 256. This implies that the frame size of SIM must be dividable by 2 in our case, to obtain integer numbers for the frame size of PALM.
- The central slice of the SIM image stack must be the focal plane later in the 3D PALM experiment. Any drift in z that occurs between the SIM acquisition and the following PALM recording must be compensated. In course of the long PALM acquisition, active drift control is essential. When matching both images in z, the PALM slice thickness must be exactly adapted to the SIM slice thickness via the 'PALM-Rendering' view tab of the ZEN software. Even then, one should be aware that in SIM the assignment of light contribution by deconvolution (DCV) will be less precise than the localization of molecules. Hence there is the chance of non-matching structures. Aligning SIM and PALM images for one plane is more reliable. In this case the SIM image plane is used for PALM recording and all localized events can be assigned to one plane.
- SIM raw data acquisition should start with the longer wavelength to minimize bleaching and cross-excitation.
- If cropping is needed during 3D-SIM raw data acquisition, crop only centrally and do not use panning, otherwise PALM and SIM images will not match later on.
- During 3D-SIM raw data acquisition the slice number has to be set to an uneven number to have the focal plane later as the central slice.
- If beads used for the correction template are on the coverslip, refractive index mismatches do not appear. If the sample is also fixed onto the coverslip the obtained correction is reliable. However, if the sample is fixed on the slide, refractive index mismatches might have an influence. Thus, the correction may be less accurate and the beads should be embedded in the same medium as the sample.
- To perform channel alignment for PALM and the co-localization of molecules, please regard that the ‘Channel Alignment’ processing function will work on PALM vector maps only if fiducial markers are present.
Recipes
- Phosphate buffered saline solution 10x concentrate (10x PBS)
1x solution is prepared by 10-fold dilution in ddH2O
Acknowledgments
We thank Andrea Kunze and Joachim Bruder for excellent technical assistance. The protocol was developed based on previously published work of Schubert (2014), Schubert and Weisshart (2015).
References
- Doležel, J., Binarová, P. and Lucretti, S. (1989). Analysis of nuclear DNA content in plant cells by flow cytometry. Biol Plant 31: 113-120.
- Jackson, D. A., Hassan, A. B., Errington, R. J. and Cook, P. R. (1993). Visualization of focal sites of transcription within human nuclei. EMBO J 12(3): 1059-1065.
- Papantonis, A. and Cook, P. R. (2013). Transcription factories: genome organization and gene regulation. Chem Rev 113(11): 8683-8705.
- Rieder, D., Trajanoski, Z. and McNally, J. G. (2012). Transcription factories. Front Genet 3: 221.
- Schubert, V. (2014). RNA polymerase II forms transcription networks in rye and Arabidopsis nuclei and its amount increases with endopolyploidy. Cytogenet Genome Res 143(1-3): 69-77.
- Schubert, V. and Weisshart, K. (2015). Abundance and distribution of RNA polymerase II in Arabidopsis interphase nuclei. J Exp Bot 66(6): 1687-1698.
- Zhao, Z. W., Roy, R., Gebhardt, J. C., Suter, D. M., Chapman, A. R. and Xie, X. S. (2014). Spatial organization of RNA polymerase II inside a mammalian cell nucleus revealed by reflected light-sheet superresolution microscopy. Proc Natl Acad Sci U S A 111(2): 681-686.
Article Information
Copyright
© 2016 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Weisshart, K., Fuchs, J. and Schubert, V. (2016). Structured Illumination Microscopy (SIM) and Photoactivated Localization Microscopy (PALM) to Analyze the Abundance and Distribution of RNA Polymerase II Molecules on Flow-sorted Arabidopsis Nuclei. Bio-protocol 6(3): e1725. DOI: 10.21769/BioProtoc.1725.
Category
Plant Science > Plant cell biology > Cell imaging
Plant Science > Plant cell biology > Organelle isolation
Cell Biology > Cell imaging > SIM
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.