- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

PhagoKinetic Track Assay: Imaging and Analysis of Single Cell Migration

Published: Vol 6, Iss 1, Jan 5, 2016 DOI: 10.21769/BioProtoc.1699 Views: 10834
Reviewed by: Guillaume Bompard Shannon RuppertAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Apr 2015
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics








Abstract
Cell migration is a highly complex and dynamic biological process, essential in several physiological phenomena and pathologies including cancer dissemination and metastasis formation. Thus understanding single cell migration is highly relevant and requires a suitable image-based assay. Depending on the speed of the moving cells, one may require fast time-lapse microscopy, which is not always suitable for high-throughput screening. To overcome this, a quantitative and fixed single cell migration assay was developed based on the PhagoKinetic Tracks (PKT) procedure. Briefly, cells are seeded on top of a monolayer of carboxylated latex beads, and as cells migrate, they phagocytose these beads and leave behind a migratory track. These bead-free migratory tracks can be visualized using a standard bright field microscope and analysed for a multiparametric quantitative assessment of single cell migration (Naffar-Abu-Amara et al., 2008).
Here we describe a detailed and optimized protocol of the PKT assay, adaptable for both RNAi and drug screening (van Roosmalen et al., 2015). This protocol allows the user to study migratory behaviour at the single cell level, without fast and live-imaging microscopy.
Materials and Reagents
- Tumour cells [in particular, this assay was set up for H1299 non-small cell lung carcinoma cells (ATCC, catalog number: CRL-5803 ) and breast cancer cell lines MDA-MB-231 (ATCC, catalog number: HBT-26 ),
- MDA-MB-417.5 (kindly provided by Joan Massagué) and Hs578T (ATCC, catalog number: HBT-126 )]
- Phosphate buffered saline (PBS) without Ca/Mg (any supplier)
- Trypsin-EDTA (Life Technologies, InvitrogenTM)
- Fibronectin from bovine plasma (Sigma-Aldrich, catalog number: F1141 )
- White carboxylate-modified latex beads (CML Latex Beads, 4% w/v, 0.4 µm), and number of particles is given in the MSDS/COA documents provided by the manufacturer (Thermo Fisher Scientific, catalog number: C37238 )
- Black 96-well flat-bottom microscopy plates (µClear 96-well plate) (Greiner Bio-One GmbH, catalog number: 655090 )
- 4% Formaldehyde (any supplier) in PBS
- RPMI medium (Life Technologies)
- Fetal Bovine Serum (FBS) (PAA Laboratories)
- Penicillin-Streptomycin (Penicillin-Streptomycin) (Life Technologies, Gibco®)
- Complete medium (see Recipes)
Equipment
- Cell culture set up, including laminar flow hood, cell culture incubator (37 °C and 5% CO2), cell counter or counting chamber, multichannel pipets
- BD Pathway 855 BioImager (BD Biosciences); or another brand or type of inverted microscope equipped with brightfield illumination, automated stage, 10x objective and software for automated imaging
- Hydroflex plate washer (Tecan Trading AG)
Software
- WIS-PhagoTracker v2_3_12 (Weizmann Institute of Science, http://www.weizmann.ac.il/vet/IC/software/wis-phagotracker; open source software)
- Montage2MRC (Weizmann Institute of Science, http://www.weizmann.ac.il/vet/IC/software/wis-phagotracker; open source software)
- KNIME (optional) (www.knime.org; open source software)
Procedure
- Day 1
- In a sterile laminar flow hood, add 40 µl/well fibronectin (10 µg/ml in PBS) to a 96-well µClear plate. Transfer the plate to a cell culture incubator and incubate for 1 h at 37 °C.
- Aspirate fibronectin without scratching the surface, and wash twice with PBS (preferentially using plate washer). Add 100 µl PBS per well and proceed to the next step.
- Swirl the stock flask of CML latex beads and aliquot 250 µl beads (approximately 3.25 x 1011 particles) per plate and spin down at 13,000 rpm for 5 min. Discard supernatant and resuspend bead pellet in 1 ml PBS. Vortex for 1 min and spin down at 13,000 rpm for 5 min. Remove supernatant and resuspend bead pellet in 1 ml PBS. Transfer resuspended beads to 15 or 50 ml tube and add PBS to a final beads suspension volume of 7 ml per plate.
- Remove PBS from fibronectin-coated plate without scratching the surface and add 70 µl beads per well.
- Visually check the plate and make sure the surface of the well is completely covered with beads and incubate for 1 h at 37 °C.
- Use plate-washer and gently wash 7 times with PBS (200 µl per well). If no plate-washer is available, carefully wash the plate manually using a multichannel and ensure the bead-coated surface is not scratched. After washing, add 100 µl PBS per well.
- Wrap the plate in parafilm and store plate at 4 °C for next day use.
- We would advise to prepare fresh beads for each experiment (as explained above) and avoid storage for long period.
- In a sterile laminar flow hood, add 40 µl/well fibronectin (10 µg/ml in PBS) to a 96-well µClear plate. Transfer the plate to a cell culture incubator and incubate for 1 h at 37 °C.
- Day 2
- Carefully aspirate PBS from bead-coated plate and replace with 100 µl complete medium.
- Prepare cells (e.g., from culture flask, or after siRNA transfection) and resuspend into single cell suspension.
- Seed 50-200 cells in a 50 µl single cell suspension (depending on cell size & migration capacity) and return plate to cell culture incubator. The final volume is 150 μl (100 µl complete medium + 50 µl single cell suspension) in which 50-200 cells will be allowed to form single-cell migration tracks.
- We recommend that cells are allowed to attach for 1 h before adding chemical compounds or inhibitors.
- Depending on migration capacity, fixate cells after 7-12 h of cell migration by removing 140 µl medium from the plate and adding 70 µl 4% formaldehyde (final formaldehyde concentration is 3.5%). Fixate for 10 min at RT.
- Wash plate twice with PBS (200 µl per well) without scratching the bead-coated surface.
- Wrap the plate in parafilm and store at 4 °C until imaging.

Figure 1. Microscopy set up of PhagoKinetic track assay. A. Non-homogenous illumination in the original brightfield image is minimized by adding a light diffuser (tip box lid), providing a clear image with equal intensities. B. A seamless montage of the largest square without cell borders (red box) allows the user to record a maximal number of complete cell tracks.
- Carefully aspirate PBS from bead-coated plate and replace with 100 µl complete medium.
- Imaging and analysis
- Use a brightfield microscope with 10x objective to visualize bead-free migratory tracks. Whole-well montages were obtained as followed:
- The transmitted light source is above the 96-plate. For equal illumination, place a tipbox lid on top of the plate to diffuse the light (see Figure 1a).
- Use laser autofocus in each well to focus before acquisition.
- Use montage-settings to acquire the largest montage of the well without imaging well-borders (example in Figure 1b).
- The transmitted light source is above the 96-plate. For equal illumination, place a tipbox lid on top of the plate to diffuse the light (see Figure 1a).
- Use Montage2MRC to convert all montages of one plate into a single r6d-file, following the software instructions.
- Run PhagoTracker software and analyse images according to the software guidelines (Figure 2a-b). The PhagoTracker user guide is very detailed and contains a myriad of information to optimize your image analysis. Briefly, the following settings were used:
- In pre-processing, set Correct Illumination to ‘Non’.
- Use a High Pass & Low Pass filter, and set the Adjust Contrast to ‘Whole Well’.
- For Track Detection, we use the recommended threshold settings. Depending on gray values, overall intensity, cell size and track size, you might need to find optimal settings.
- Use Track Filtering to reject tracks without cells and to reject tracks with more than 1 cell. Briefly, the software detects bright objects (migratory paths) and dark objects (cells). It subsequently determines whether each migratory path is accepted (1 path with 1 cell) or rejected (1 path with 0 cells, or 1 path with >1 cells). Tracks can also be manually accepted or rejected after analysis. This is not necessary if the image analysis parameters are set correctly.
- In pre-processing, set Correct Illumination to ‘Non’.
- PhagoTracker calculates multiple morphometric features for each migratory track, including Total Area, Net Area, Minor Axis, Major Axis, Axial Ratio and Perimeter as measurements of track size and shape. Furthermore, the Solidity and Roughness of each track are calculated, which represent membrane activity and ruffling during migration (Figure 2c-d). The software generates 2 files with quantitative output: one file contains data of all accepted individual tracks, the other contains average values and standard deviations of accepted, rejected and all tracks per well.
- Quantitative output can be further analysed using PhagoTracker (see user guide), or processed using preferred programs, like Excel, Graphpad, R or KNIME.
- To automate your analysis, KNIME is recommended. This open-source software contains a plethora of ready-to-use analysis nodes, including a plate lay out joiner, different normalization methods (Z-score, B-score, NPI, POC, etc.) and plotting tools (2D/3D scatter plot, etc.), to build an analysis pipeline.
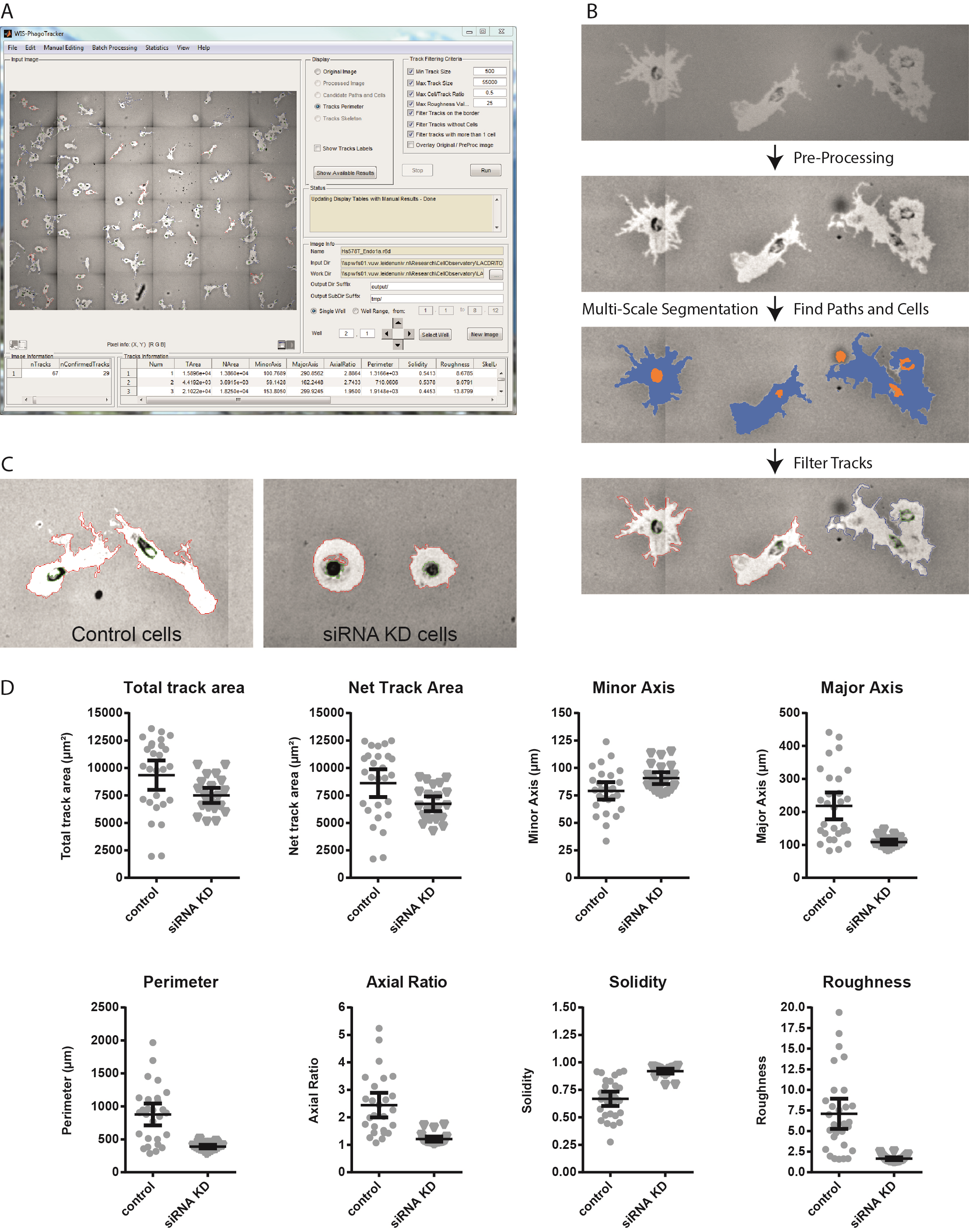
Figure 2. PhagoTracker software and image analysis procedure. A. The PhagoTracker software interface. B. PhagoTracker software performs image analysis in several steps. Images are pre-processed to enhance contrast and correct illumination. Multi-Scale segmentation finds candidate cell and path segments (shown in orange and blue, respectively) which are subsequently combined into track segments. The candidate tracks are filtered based on the selection criteria and are marked as confirmed (in red) or rejected (in blue). C. Representative images of Hs578T breast cancer cells. Control cells show large elongated tracks with a rough border. Cells with siRNA knockdown (KD) do not migrate and form round tracks. D. Quantification of individual tracks shown in C. Solidity is calculated as (track area/ area of the convex hull) and roughness as (perimeter2) / (4π*Total Area).
- To automate your analysis, KNIME is recommended. This open-source software contains a plethora of ready-to-use analysis nodes, including a plate lay out joiner, different normalization methods (Z-score, B-score, NPI, POC, etc.) and plotting tools (2D/3D scatter plot, etc.), to build an analysis pipeline.
- Use a brightfield microscope with 10x objective to visualize bead-free migratory tracks. Whole-well montages were obtained as followed:
Notes
Important considerations
- Concerning the plate preparation:
- There is a very small, negligible, risk that you wash away the beads, but this only occurs if you use a lot of force. In reality, this problem never occurs.
- It is very easy to gently tap or shake (horizontally) the plate to distribute the beads solution and cover the surface of the wells. This protocol uses an excess of beads and beads are coated by sedimentation, so the coverage is very uniform.
- There is a very small, negligible, risk that you wash away the beads, but this only occurs if you use a lot of force. In reality, this problem never occurs.
- At all times, do not scratch the bottom of the plate. Scratches in the bead-coating can appear as migratory tracks and could affect your analysis. In general, minor/small scratches can be ignored, as they do not affect the analysis. Indeed, in case a scratch is detected as track, there is no cell in it and therefore it would be rejected.
- This protocol can be used for other cell lines, however we recommend a pilot experiment to find optimal conditions for any cell line used. These include:
- Size, type and surface charge of the beads. Different sizes, types and charge of beads have been tested before and are clearly described by Naffar-Abu-Amara et al. Briefly, CML latex beads with a size of 340 or 400 nm work best. Beads with a low charge density will interact strongly with the surface of the well and consequently cells cannot remove the beads as they migrate. The beads used in this protocol have a charge density of 85-100 µEq/g and can be used for a wide range of cells. The catalog number is provided in the materials and reagents section allowing users to order the same beads.
- Optimal cell density, which is dictated by several parameters such as cell size (the larger the cells, the lower the density) and cell motility (the higher the speed, the lower the cell density).
- Time of migration. Less motile cell lines might require longer incubation time for migratory track formation.
Aim for a maximum number of bead-free migratory tracks at the end of the assay that do not intersect. Track intersection can be inspected visually. You see 1 bright track and 2 cells, indicating either intersection or cell division (Figure 2b, first panel). The PhagoTracker software saves the path (migratory tracks) and cells image that is generated during analysis (Figure 2b, third panel). You can decide whether tracks are automatically rejected or not, and which criteria to use for rejection.
- Size, type and surface charge of the beads. Different sizes, types and charge of beads have been tested before and are clearly described by Naffar-Abu-Amara et al. Briefly, CML latex beads with a size of 340 or 400 nm work best. Beads with a low charge density will interact strongly with the surface of the well and consequently cells cannot remove the beads as they migrate. The beads used in this protocol have a charge density of 85-100 µEq/g and can be used for a wide range of cells. The catalog number is provided in the materials and reagents section allowing users to order the same beads.
Recipes
- Complete medium
RPMI medium was supplemented with 10% fetal bovine serum and 25 IU/ml penicillin and 25 µg/ml streptomycin.
Acknowledgments
This protocol was developed based on Naffar-Abu-Amara et al. (2008) and further optimized in the Division of Toxicology, Leiden Academic Centre for Drug Research, Leiden University. The work was funded by grants from the EU-FP7 - Systems Microscopy NoE (grant no. 258068 to B. van de Water) and the Dutch Cancer Society (UL2007-3860).
References
- Naffar-Abu-Amara, S., Shay, T., Galun, M., Cohen, N., Isakoff, S. J., Kam, Z. and Geiger, B. (2008). Identification of novel pro-migratory, cancer-associated genes using quantitative, microscopy-based screening. PLoS One 3(1): e1457.
- Sharon, E., Galun, M., Sharon, D., Basri, R. and Brandt, A. (2006). Hierarchy and adaptivity in segmenting visual scenes. Nature 442(7104): 810-813.
- van Roosmalen, W., Le Devedec, S. E., Golani, O., Smid, M., Pulyakhina, I., Timmermans, A. M., Look, M. P., Zi, D., Pont, C., de Graauw, M., Naffar-Abu-Amara, S., Kirsanova, C., Rustici, G., Hoen, P. A., Martens, J. W., Foekens, J. A., Geiger, B. and van de Water, B. (2015). Tumor cell migration screen identifies SRPK1 as breast cancer metastasis determinant. J Clin Invest 125(4): 1648-1664.
Article Information
Copyright
© 2016 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Fokkelman, M., Roosmalen, W. V., Rogkoti, V., Le Dévédec, S. E., Geiger, B. and Water, B. V. D. (2016). PhagoKinetic Track Assay: Imaging and Analysis of Single Cell Migration. Bio-protocol 6(1): e1699. DOI: 10.21769/BioProtoc.1699.
Category
Cancer Biology > General technique > Cell biology assays > Cell migration
Cancer Biology > Invasion & metastasis > Cell biology assays > Cell migration
Cell Biology > Cell imaging > Live-cell imaging
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.