- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Chromatin Immunoprecipitation (ChIP) Assay for Detecting Direct and Indirect Protein – DNA Interactions in Magnaporthe oryzae


Published: Vol 5, Iss 21, Nov 5, 2015 DOI: 10.21769/BioProtoc.1643 Views: 16026
Reviewed by: Fanglian HeArsalan Daudi
Original research article
The authors used this protocol in:

Oct 2014
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics









Abstract
Chromatin immunoprecipitation (ChIP) is a powerful technology for analyzing protein-DNA interactions in cells. Robust ChIP procedures have been established for investigating direct interactions between protein and DNA. However, detecting indirect protein-DNA interactions in vivo is challenging. Recently, we used ChIP to analyze an indirect protein-DNA interaction between a putative histone demethylase, MoJmjC, and the promoter of the superoxide dismutase 1-encoding gene MoSOD1 in the rice blast fungus Magnaporthe oryzae (M. oryzae) (Fernandez et al., 2014). We tagged MoJmjC with the 3x FLAG epitope (Fernandez et al., 2014), instead of the larger and more commonly used GFP epitope, to mitigate against steric hindrance. We also employed a two-step cross-linking strategy using DSG and formaldehyde-rather than the one-step formaldehyde cross-linking procedure more frequently employed for analyzing direct protein-DNA interactions - in order to better capture the indirect MoJmjC-MoSOD1 DNA interactions in vivo. In addition, we have shown that two-step cross-linking is suitable for ChIP analysis of direct protein-DNA interactions between a GATA transcription factor, Asd4, and its cognate binding site (Marroquin-Guzman and Wilson, 2015). Here, we provide a detailed protocol for chromatin immunoprecipitation, with versatile two-step cross-linking, in M. oryzae.
Materials and Reagents
- Oak Ridge Centrifuge Tubes (Thermo Fisher Scientific, NalgenetmTM, catalog number: 3118-0050 )
- Miracloth (EMD Millipore Corporation, catalog number: 475855-1R )
- Saccharomyces cerevisiae strain XK1-25: MATa trp1 (kindly provided by Dr. Jin-Rong Xu, Dept. of Botany and Plant Pathology, Purdue University)
- pHZ126 yeast shuttle vector (kindly provided by Dr. Jin-Rong Xu, Dept. of Botany and Plant Pathology, Purdue University, USA)
- QIAquick gel extraction kit (QIAGEN, catalog number: 28704 )
- Alkali-cation yeast transformation kit (MP Biomedicals, catalog number: 2200-200 )
- QIAprep Spin Miniprep Kit (QIAGEN, catalog number: 27104 )
- UltraClean Midi Prep Kit (MO BIO Laboratories, catalog number: 12700-20 )
- Zymoprep Yeast Plasmid Mini Prep II Kit (Epigenetics, catalog number: D2004 )
- Formaldehyde (Thermo Fisher Scientific, PierceTM, catalog number: 28906 )
- DSG (Thermo Fisher Scientific, catalog number: 20593 )
- CelLyticTM PN Plant Nuclear Isolation Kit (Sigma-Aldrich, catalog number: CELLYTPN1 )
- Nuclei isolation buffer (NIB) (included in the CelLyticTM PN Plant Nuclear Isolation Kit)
- Triton X-100 Solution (10% in H2O) (MBL International, catalog number: JM-2104-100 )
- Proteinase inhibitor cocktail (Sigma-Aldrich, catalog number: P8215 )
- Deoxycholate (Sigma-Aldrich, catalog number: D6750 )
- Sodium butyrate (Sigma-Aldrich, catalog number: B5887 )
- Sepharose 4B beads (Sigma-Aldrich, catalog number: CL4B200 )
- ANTI-FLAG® M2 Affinity Gel (Sigma-Aldrich, catalog number: A2220 )
- Anti-IgG agarose (Sigma-Aldrich, catalog number: A0919 )
- 3x FLAG® Peptide (Sigma-Aldrich, catalog number: F4799 )
- Glycine (Thermo Fisher Scientific, InvitrogenTM, catalog number: 15527-013 )
- Proteinase K (Thermo Fisher Scientific, catalog number: EO0491 )
- RNase A (Thermo Fisher Scientific, catalog number: EN0531 )
- Wizard PCR Clean-up kit (Promega Corporation, catalog number: A9281 )
- Yeast nitrogen base without amino acids (Difco, catalog number: 0919-15 )
- Trp DO supplement (Clontech, catalog number: 630413 )
- Disuccinimidyl glutarate (DSG) (Thermo Fisher Scientific, catalog number: 20593 )
- Lysing Enzymes from Trichoderma harzianum (Sigma-Aldrich, catalog number: L1412 )
- Hygromycin B (Thermo Fisher Scientific, GibcoTM, catalog number: 10-687-010 )
- SD-Trp solid medium (see Recipes)
- SD-Trp liquid solution (see Recipes)
- LB agar plates supplemented with 100 μg/ml ampicillin (see Recipes)
- Liquid complete medium (see Recipes)
- DSG cross-linking buffer (see Recipes)
- 1x NIB (see Recipes)
- NIBA (see Recipes)
- Nuclear lysis buffer (see Recipes)
- HEPES-buffered saline (HBS) (see Recipes)
- 0.1 M glycine HCl (pH 3.5) (see Recipes)
- Elution buffer (see Recipes)
- OM buffer (see Recipes)
- ST buffer (see Recipes)
- STC buffer (see Recipes)
- PTC buffer (see Recipes)
- Bottom medium (see Recipes)
- Top medium (see Recipes)
Equipment
- Benchtop shakers (Eppendorf, catalog number: M1354-0000 )
- VirTis VirSonic 50 Ultrasonic Cell Disrupter Disruptor (VirTis company)
- Centrifuge (Eppendorf, model: 5415D )
- Mini-Tube Rotators (Thermo Fisher Scientific, Fisher Scientific, catalog number: 05-450-127 )
Procedure
- The first step towards ChIP analysis in M. oryzae is to construct a plasmid expressing the target gene fused to 3x FLAG from its native promoter. We routinely use the yeast shuttle vector and methodology described by Zhou et al. (2011), summarized below:
- Vector preparation
A yeast shuttle vector, pHZ126 (Zhou et al., 2011), is available for generating C-terminal 3x FLAG fusion constructs. In addition to 3x FLAG encoding sequences, pHZ126 carries TRP1 for selection in yeast Trp- strains, AmpR for selection in Escherichia coli (E. coli) strains, and HygR for selection based on hygromycin resistance in M. oryzae. In preparation for subcloning the target gene, 100 μg of pHZ126 vector DNA is linearized by digestion with XhoI and, after size confirmation on a 0.8% agarose gel, purified with the QIAquick gel extraction kit, following the manufacturer’s protocol. - Target gene amplification.
- The following primer pairs are used to generate the target gene PCR product with overlapping sequences for fusing in-frame with the 3xFLAG coding sequence carried on the pHZ126 shuttle vector (Zhou et al., 2011):
Gene-fw: 5’-TATAGGGCGAATTGGGTACTCAAATTGGTTXXXXXXXXXXX-3’
Gene-rv: 5’-TTTATAATCACCGTCATGGTCTTTGTAGTCYYYYYYYYYYY-3’
Note: The underlined nucleotide sequences are specific for the target gene. The non-underlined sequences are used for homologous recombination onto the yeast shuttle vector pHZ126. In order to generate an in-frame gene fusion between target gene and the 3xFLAG tag sequence on pHZ126, the nucleotide sequences in italics in the primer Gene-rv encode the last codon before the target gene stop codon. For best results, the gene specific sequences are designed with a melting temperature of 65 °C, an ideal GC content of 40-60%, and 35-40 cycles of the PCR reaction should be performed. - The target gene amplicon is gel purified using the QIAquick gel extraction kit, following the manufacturer’s protocol.
- The following primer pairs are used to generate the target gene PCR product with overlapping sequences for fusing in-frame with the 3xFLAG coding sequence carried on the pHZ126 shuttle vector (Zhou et al., 2011):
- The target gene PCR product from step A2 is subcloned into the XhoI digested yeast shuttle vector pHZ126 by co-transformation into the yeast strain XK1-25, following the protocol of the Alkali-Cation Yeast Transformation Kit.
Note: The non-underlined nucleotide sequences added to the Gene-fw and Gene-rv primers above facilitate recombination onto pHZ126 by aligning with vector sequences on either side of the XhoI restriction site. Thus, the PCR product does not need to be digested with XhoI.
Screening of yeast transformants carrying the target gene-3x FLAG fusion in pHZ126 is performed on plates containing SD-Trp solid medium followed by colony PCR with gene-specific primer pairs for verification. - Positive yeast colonies are propagated on a fresh SD-Trp plate, and yeast plasmid DNA is extracted using the Zymoprep Yeast Plasmid Mini Prep II kit.
- The yeast plasmid DNA is then transformed into competent cells of E. coli strain DH5a, using standard procedures. E. coli cells carrying the yeast plasmid DNA are selected on ampicillin-containing LB agar plates, followed by verification using colony PCR.
- The plasmid DNA is extracted from E. coli using the Mo Bio UltraClean Midi Plasmid Prep kit, and an aliquot is sequenced to ensure the fidelity of the target gene sequence and 3x FLAG fusion.
- Vector preparation
- The next step towards ChIP is to integrate the target gene fused to 3x FLAG into the M. oryzae genome. This is achieved by transforming the plasmid DNA isolated in step A6 into M. oryzae protoplasts and selecting for hygromycin resistance. Protoplast generation and transformation, as well as screening for fungal transformants, are performed as described previously (Fernandez et al., 2012; Wilson et al., 2010) and summarized as follows:
- Mycelia are grown in 350 ml of liquid complete medium with continuous shaking at 150 rpm for 48 h at 24 °C, harvested with sterile Miracloth and washed with sterile distilled water.
- The mycelia are divided into two parts, and each is incubated in 40 ml of OM buffer at 29 °C with shaking at 75 rpm.
- The mycelia/OM solution is aliquoted into two Oak Ridge centrifuge tubes, and overlaid with the same volume of cold ST buffer on ice. After centrifugation at 5,000 rpm for 15 min at 4 °C in a swinging bucket rotor, protoplasts are recovered from the OM/ST interface using a 1 ml sterile pipette and pooled.
- After gently mixing with the same volume of cold STC buffer, the protoplasts are pelleted by centrifugation at 3,000 rpm for 10 min at 4 °C, followed by three washes in 20 ml of cold STC buffer.
- The protoplast pellet is resuspended in cold STC buffer to give a final concentration of 2-5 x 108 protoplasts/ml.
- 100 ul of protoplasts (~1 x 107) are mixed with 5 μg of plasmid DNA, and incubated at room temperature for 20 min. Protoplasts and DNA are then gently mixed with 1 ml of PTC buffer by gently inverting the tube, and the solution is incubated at room temperature for a further 20 min.
- To select for transformants, the protoplast/PTC solution is next mixed with 100 ml of cool but molten (45 °C) bottom medium and poured into four plates (~25 ml/plate). The plates are allowed to set, then incubated in the dark for at least 24 h at 24 °C. The bottom medium is then overlaid with the same volume of top medium containing 200 μg/ml Hygromycin B, and the plates are incubated under dark conditions until colonies appear (approx. 2 -3 weeks).
- Hygromycin resistant colonies are screened for target gene-3x FLAG genomic integration by PCR using gene-specific primer pairs. Expression of the target gene can be further verified by quantitative real time PCR (qRT-PCR) and immunoblot analysis using the ANTI-FLAG antibody.
- Mycelia are grown in 350 ml of liquid complete medium with continuous shaking at 150 rpm for 48 h at 24 °C, harvested with sterile Miracloth and washed with sterile distilled water.
- Preparation of fungal mycelia for ChIP
- Once M. oryzae strains that express the target gene fused to 3x FLAG have been verified, these can then be grown up for ChIP analysis. Fungal strains carrying the target gene-3x FLAG fusion are grown on agar plates for 5-10 days before the leading edge of the plate colony is excised and blended with 350 ml liquid complete medium (CM). The mycelia and CM mix is decanted into a suitable flask and the fungus is allowed to grow at 24 °C for 48 h with continuous shaking at 150 rpm (Figure 1A). For some studies, mycelia might be transferred to a second, defined, minimal medium for 16 h of additional growth. A wild type strain or a strain carrying the empty pHZ126 vector should be used as the experimental control.
- For each strain, fungal mycelia are collected using two layers of Miracloth and the harvested mycelia are washed extensively with sterile water (Figure 1B-C).

Figure 1. Collection of fungal mycelia. A. Fungal strains carrying the target gene-3x FLAG fusion are grown in liquid complete medium at 24 °C. B. After 48 h, the mycelia are harvested by filtering the culture through two pieces of Miracloth placed in a funnel. The mycelium is washed with sterile water. C. The mycelial mat is gently patted with paper towels to remove excess water.
- Once M. oryzae strains that express the target gene fused to 3x FLAG have been verified, these can then be grown up for ChIP analysis. Fungal strains carrying the target gene-3x FLAG fusion are grown on agar plates for 5-10 days before the leading edge of the plate colony is excised and blended with 350 ml liquid complete medium (CM). The mycelia and CM mix is decanted into a suitable flask and the fungus is allowed to grow at 24 °C for 48 h with continuous shaking at 150 rpm (Figure 1A). For some studies, mycelia might be transferred to a second, defined, minimal medium for 16 h of additional growth. A wild type strain or a strain carrying the empty pHZ126 vector should be used as the experimental control.
- Two-step cross-linking [modified from Nowak et al. (2005)] of fungal mycelia for ChIP
- First step cross-linking: 1 g of washed mycelia is suspended in 100 ml of DSG cross-linking buffer and incubated for 45 min at room temperature with continuous shaking at 100 rpm.
- Second step cross-linking: formaldehyde is added to a final concentration of 1% for the last 20 min of cross-linking with DSG.
- Terminate cross-linking: glycine is added to a final concentration of 0.125 M, and incubated at room temperature for 10 min.
- The mycelia are harvested with Miracloth, washed excessively with sterile distilled water, flash frozen in liquid nitrogen and stored at -80 °C until use.
- First step cross-linking: 1 g of washed mycelia is suspended in 100 ml of DSG cross-linking buffer and incubated for 45 min at room temperature with continuous shaking at 100 rpm.
- Isolation of crosslinked chromatin with CelLyticTM PN Plant Nuclear Isolation Kit, following the manufacturer’s protocol
Note: From procedure E to F, perform all steps at 4 °C. All buffers and equipment used should be pre-cooled to 4 °C.- Crosslinked mycelia are ground to a fine powder in liquid nitrogen using a pre-chilled mortar and pestle.
- 100 mg of mycelial powder is suspended in 500 μl of 1x NIB in a 1.5 ml Eppendorf tube by vigorous vortexing.
- The suspension is filtered through 2 layers of sterile Miracloth into a fresh 1.5 ml Eppendorf tube.
- The suspension is centrifuged at 1,260 x g for 10 min at 4 °C.
- The supernatant is discarded and the pellet is re-suspended in 500 μl of NIBA.
- For the lysis of cell membranes, 15 μl of 10% Triton X-100 is added to a final concentration of 0.3% and the solution is gently inverted to mix.
- The solution is centrifuged at 12,000 x g for 5 min at 4 °C.
- The supernatant is removed and the pellet resuspended in 500 μl of NIBA.
- The solution is centrifuged at 12,000 x g for 5 min at 4 °C, and the supernatant discarded.
- The resulting pellet is the crude nuclear fraction.
- Crosslinked mycelia are ground to a fine powder in liquid nitrogen using a pre-chilled mortar and pestle.
- Chromatin shearing by sonication
- The crude nuclear fraction from step E10 is resuspended in 300 μl of pre-cooled nuclear lysis buffer.
- Chromatin is extracted from nuclei and sheared using the VirTis VirSonic 50 Ultrasonic Cell Disrupter Disruptor. Samples are kept on ice throughout the procedure. To generate chromatin fragments in the optimal size range of 200 bp-1,000 bp (with an average fragment size of 500 bp), a Microtip power setting of 4 in cycles of 20 sec “on” and 20 sec “off” during a total treatment time of 6 min can be used for M. oryzae nuclei.
- Samples are centrifuged at 12,000 x g for 5 min at 4 °C, and the supernatant containing the nuclear lysate and sheared chromatin is transferred to a fresh tube.
- The crude nuclear fraction from step E10 is resuspended in 300 μl of pre-cooled nuclear lysis buffer.
- Chromatin immunoprecipitation
- The nuclear lysate containing sheared chromatin is precleared by incubating with 30 μl Sepharose 4B beads with gentle rotation for 4 h at 4 °C.
- The beads are centrifuged at 5,000 x g for 30 sec at 4 °C.
- The supernatant is collected into 3 x 100 μl aliquots in fresh Eppendorf tubes. One sample is saved until step H1 to serve as the input chromatin control. All samples are placed on ice until use.
- The affinity resin is prepared according to the manufacturer's instructions.
- Briefly, 20 µl ANTI-FLAG M2 Affinity Gel is removed to a sterile 1.5 ml tube. The gel is washed with 0.5 ml HBS twice followed by 0.5 ml 0.1 M glycine HCl (pH 3.5) once. This is followed by three washes with 0.5 ml HBS. Between washing steps the resin is pelleted by spinning at 5,000 x g for 30 sec at 4 °C.
- 20 µl Anti-IgG Agarose resin is prepared in the same way as above and is used to determine the background of non-specific chromatin binding to the gel.
- Briefly, 20 µl ANTI-FLAG M2 Affinity Gel is removed to a sterile 1.5 ml tube. The gel is washed with 0.5 ml HBS twice followed by 0.5 ml 0.1 M glycine HCl (pH 3.5) once. This is followed by three washes with 0.5 ml HBS. Between washing steps the resin is pelleted by spinning at 5,000 x g for 30 sec at 4 °C.
- 100 µl of cleared nuclear lysate from Step G3 is placed in separate tubes containing the ANTI-FLAG or Anti-IgG agarose resin and incubated overnight at 4 °C with gentle agitation.
- The resin is washed four times in 0.5 ml HBS buffer containing 0.1% TritonX-100. Following these wash steps the beads are pelleted by spinning at 5,000 x g for 30 sec at 4 °C.
- To elute the protein-DNA complex, 100 μl Elution buffer is added to the resin pellet and incubated for 30 min at 4 °C with gentle agitation.
- The sample is centrifuged at 5,000 x g for 30 sec at 4 °C and the supernatant transferred to a fresh tube.
- The nuclear lysate containing sheared chromatin is precleared by incubating with 30 μl Sepharose 4B beads with gentle rotation for 4 h at 4 °C.
- Reverse cross-linking
Note: In addition to the eluate from the ANTI-FLAG and Anti-IgG agarose resin, the input chromatin saved at step G3 is also included in the following steps.- To digest proteins, Proteinase K is added to each sample at a final concentration of 0.2 mg/ml and the solution is incubated at 45 °C for 2 h.
- To reverse the covalent association of the epitope tagged protein with its direct or indirect DNA target, NaCl is added to a final concentration of 0.2 M and the solution is incubated at 65 °C overnight.
- To digest proteins, Proteinase K is added to each sample at a final concentration of 0.2 mg/ml and the solution is incubated at 45 °C for 2 h.
- Analysis of ChIPed DNA
- Contaminating RNA is removed from the ChIPed sample by incubating with RNase A (final concentration is 0.1 mg/ml) at room temperature for 30 min.
- DNA is purified using the Wizard PCR Clean-up kit and eluted into 30 μl ddH2O.
- Quantitative real-time PCR (qPCR) is used to determine if known or suspected DNA target sequences are enriched in samples following ChIP. Using specific primers for the gene of interest (i.e., MoSOD1 in Figure 2), DNA enrichment values following ChIP are determined by subtracting the background signal of the Anti-IgG agarose treated sample from the ChIP ANTI-FLAG signal, and normalizing against the input control. For example, Fernandez and associates (2014) demonstrated that MoJmjC physically associates with the MoSOD1 promoter in a MoSIR2 dependent manner (Figure 2) such that MoSOD1 DNA is more highly enriched following ChIP by JmjCFLAG in a ∆sir2 mutant background than following ChIP by JmjCFLAG in a wild type (WT) background.
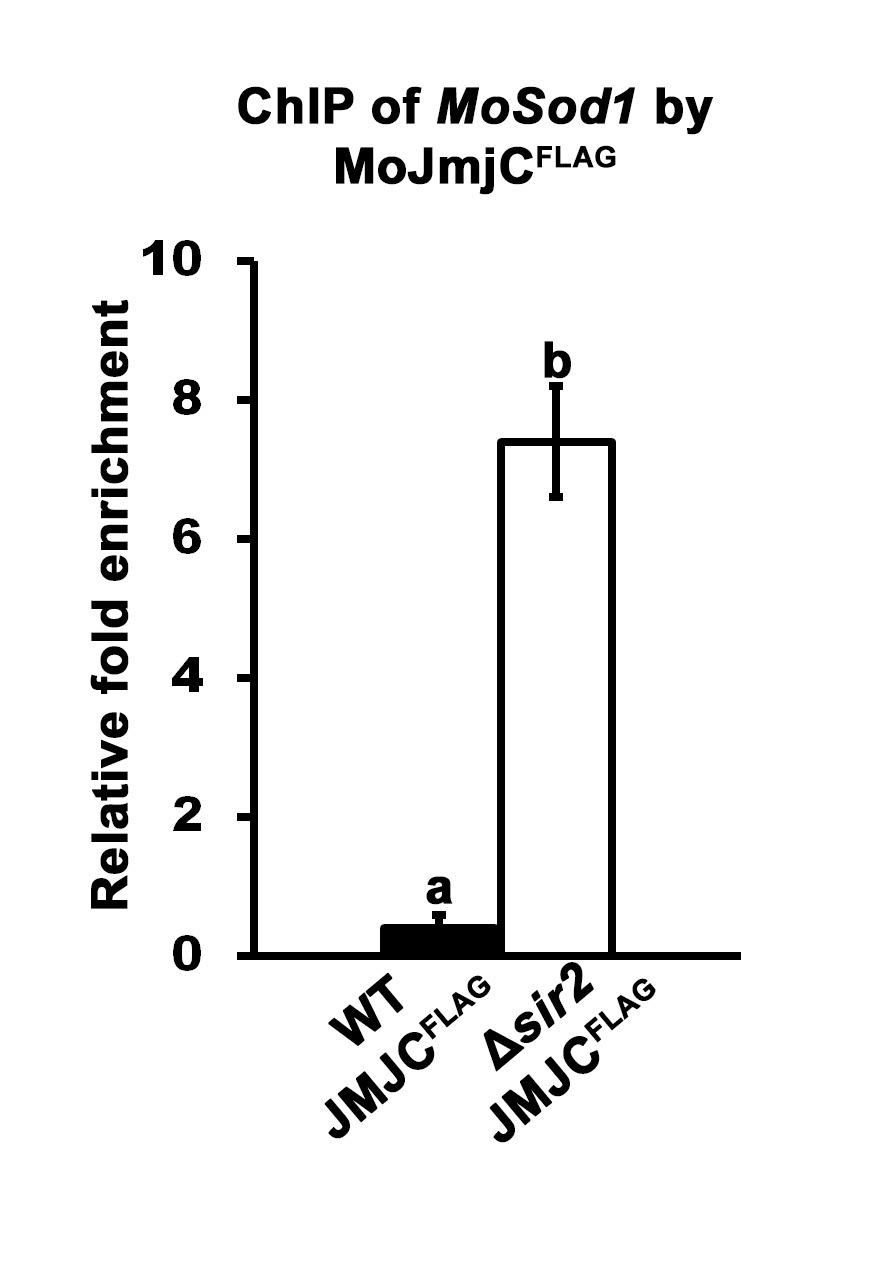
Figure 2. Quantification of ChIPed DNA. Three biological replications were performed per strain. Indirect binding of MoJmjC to MoSod1 DNA was demonstrated by ChIP following two-step cross-linking using Guy11 wild type (WT) and ∆sir2 deletion strains carrying the JMJCFLAG allele. The ANTI-FLAG M2 Affinity Gel was used in parallel with the Mouse Anti-IgG agarose. The quantification of eluted MoSod1 DNA was performed at least in triplicate by qPCR using specific primers designed in the MoSod1 promoter region (Fernandez et al., 2014). qPCR values obtained from ANTI-FLAG M2 Affinity Gel immunoprecipitation were adjusted for non-specific chromatin binding and precipitation using Anti-IgG agarose values and normalized against the input controls. The fold enrichment of MoSod1 DNA in WT JMJCFLAG ChIP samples relative to ∆sir2 JMJCFLAG ChIP samples is shown. Error bars are the standard deviation. Bars with different letters are significantly different (Student’s t-test P ≤ 0.05).
- Contaminating RNA is removed from the ChIPed sample by incubating with RNase A (final concentration is 0.1 mg/ml) at room temperature for 30 min.
Recipes
- SD-Trp solid medium
1 M sorbitol
0.67% (w/v) yeast nitrogen base without amino acids
0.074% (w/v) Trp DO supplement
1.5% agar
2% (w/v) glucose
Sterilized by autoclaving at 121 °C for 20 min
Note: Filter sterilized glucose should be added after autoclaving at a temperature below 55 °C. - SD-Trp liquid solution
1 M sorbitol
0.67% (w/v) yeast nitrogen base without amino acids
0.074% (w/v) Trp DO supplement
2% (w/v) glucose
Sterilize by autoclaving at 121 °C for 20 min
Note: Filter sterilized glucose should be added after autoclaving at a temperature below 55 °C. - LB agar plates supplemented with 100 μg/ml ampicillin
1% bacto tryptone
0.5% yeast extract 1% NaCl
1.5% bacto agar
Sterilized by autoclaving at 121 °C for 20 min, and ampicillin is added when the liquid has cooled below 55 °C - Liquid complete medium
10 g/L glucose
2 g/L peptone
1 g/L yeast extract
1 g/L casamino acids
0.1% (v/v) trace elements
0.1% (v/v) vitamin supplement
6 g/L NaNO3
0.5 g/L KCl
0.5 g/L MgSO4
1.5 g/L KH2PO4
Adjust PH 6.5
Sterilized by autoclaving at 121 °C for 30 min - DSG cross-linking buffer
20 mM HEPES (pH 7.4)
1 mM EDTA
1 mM phenylmethylsulfonyl fluoride (PMSF)
2 mM disuccinimidyl glutarate
DSG cross-linking buffer should be freshly prepared. HEPES and EDTA are sterilized by autoclaving at 121 °C for 20 min. PMSF and DSG should be added just before use. PMSF is dissolved in isopropanol or ethanol. DSG is dissolved in DMSO to make a stock solution of 0.5 M for immediate use - 1x NIB
1x NIB is diluted from 4x NIB with deionized H2O. Then DTT is added to a final concentration of 1 mM. 4x NIB is provided with the CelLyticTM PN Plant Nuclear Isolation Kit
Note: The DTT should be added just before use. - NIBA
Add proteinase inhibitor cocktail with a ratio of 1:100 to 1x NIB
Note: The proteinase inhibitor cocktail should be added just before use. - Nuclear lysis buffer
50 mM HEPES (pH 7.5)
150 mM NaCl
1 mM EDTA
1% Triton X-100
0.1% deoxycholate
0.1% SDS
10 mM sodium butyrate
1 mM PMSF
1% (v/v) proteinase inhibitor cocktail
Sterilized by autoclaving at 121 °C for 30 min
PMSF and the proteinase inhibitor cocktail should be added just before use - HEPES-buffered saline (HBS)
10 mM HEPES (pH 7.5)
150 mM NaCl
Sterilized by autoclaving at 121 °C for 30 min - 0.1 M glycine HCl (pH 3.5)
0.1 M glycine dissolved in ddH2O
Adjust pH with HCl to 3.5 - Elution buffer
50 mM Tris-HCl (pH 7.5)
200 μg/ml 3x FLAG peptide
150 mM NaCl
Dissolve 3x FLAG peptide in autoclave sterilized 1x TBS (PH 7.5) solution, and store at -20 °C - OM buffer
1.2M MgSO4
10 mM NaPO4 (pH 5.8)
0.75% (w/v) glucanex
Adjust pH 5.5 with 1 M Na2HPO4, and filter sterilize
OM buffer should be prepared fresh - ST buffer
0.6 M sorbitol
0.1 M Tris-HCl (pH 7.0)
Sterilized by autoclaving at 121 °C for 30 min - STC buffer
1.2 M sorbitol
0.1 M Tris-HCl (pH 7.5)
10 mM CaCl2
Sterilized by autoclaving at 121 °C for 30 min - PTC buffer
60% PEG 4000
10 mM Tris-HCl (pH 7.5)
10 mM CaCl2
Filter sterilize
PTC buffer should be prepared freshly. - Bottom medium
10 g/L glucose
273 g/L sucrose
2 g/L peptone
1 g/L yeast extract
1 g/L casamino acids
0.1% (v/v) trace elements
0.1% (v/v) vitamin supplement
6 g/L NaNO3
0.5 g/L KCl
0.5 g/L MgSO4
1.5 g/L KH2PO4
1.5% agar
Adjust PH 6.5 and sterilize by autoclaving at 121 °C for 20 min - Top medium
10 g/L glucose
2 g/L peptone
1 g/L yeast extract
1 g/L casamino acids
0.1% (v/v) trace elements
0.1% (v/v) vitamin supplement
6 g/L NaNO3
0.5 g/L KCl
0.5 g/L MgSO4
1.5 g/L KH2PO4
1% agar
Adjust PH 6.5 and sterilize by autoclaving at 121 °C for 20 min
Acknowledgments
This work was supported by the National Science Foundation (IOS-1145347) and USDA-NIFA (2014-67013-21559). This protocol was adapted and modified from Fernandez et al. (2014).
References
- Fernandez, J., Marroquin-Guzman, M., Nandakumar, R., Shijo, S., Cornwell, K. M., Li, G. and Wilson, R. A. (2014). Plant defence suppression is mediated by a fungal sirtuin during rice infection by Magnaporthe oryzae. Mol Microbiol 94(1): 70-88.
- Fernandez, J., Wright, J. D., Hartline, D., Quispe, C. F., Madayiputhiya, N. and Wilson, R. A. (2012). Principles of carbon catabolite repression in the rice blast fungus: Tps1, Nmr1-3, and a MATE-family pump regulate glucose metabolism during infection. PLoS Genet 8(5): e1002673.
- Marroquin-Guzman, M. and Wilson, R. A. (2015). GATA-dependent glutaminolysis drives appressorium formation in Magnaporthe oryzae by suppressing TOR inhibition of cAMP/PKA signaling. PLoS Pathog 11(4): e1004851.
- Nowak, D. E., Tian, B. and Brasier, A. R. (2005). Two-step cross-linking method for identification of NF-kappaB gene network by chromatin immunoprecipitation. Biotechniques 39(5): 715-725.
- Wilson, R. A., Gibson, R. P., Quispe, C. F., Littlechild, J. A. and Talbot, N. J. (2010). An NADPH-dependent genetic switch regulates plant infection by the rice blast fungus. Proc Natl Acad Sci U S A 107(50): 21902-21907.
- Zhou, X., Li, G. and Xu, J. R. (2011). Efficient approaches for generating GFP fusion and epitope-tagging constructs in filamentous fungi. Methods Mol Biol 722: 199-212.
Article Information
Copyright
© 2015 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Li, G., Marroquin-Guzman, M. and Wilson, R. A. (2015). Chromatin Immunoprecipitation (ChIP) Assay for Detecting Direct and Indirect Protein – DNA Interactions in Magnaporthe oryzae. Bio-protocol 5(21): e1643. DOI: 10.21769/BioProtoc.1643.
Category
Microbiology > Microbial biochemistry > Protein > Interaction
Biochemistry > Protein > Immunodetection > ChIP
Molecular Biology > DNA > DNA-protein interaction
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.