- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Computational Identification of MicroRNA-targeted Nucleotide-binding Site-leucine-rich Repeat Genes in Plants




(*contributed equally to this work) Published: Vol 5, Iss 21, Nov 5, 2015 DOI: 10.21769/BioProtoc.1637 Views: 11514
Reviewed by: Tie LiuAlberto Carbonell
Original research article
The authors used this protocol in:

Sep 2014
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
Plant genomes harbor dozens to hundreds of nucleotide-binding site-leucine-rich repeat (NBS-LRR, NBS for short) type disease resistance genes (Shao et al., 2014; Zhang et al., 2015). Proper regulation of these genes is important for normal growth of plants by reducing unnecessary fitness costs in the absence of pathogen infection. Recent studies have revealed that microRNAs are involved in regulation of NBS genes in plants (Zhai et al., 2011; Shivaprasad et al., 2012). This protocol describes computational methods for the genome-wide identification of plant NBS genes potentially regulated by microRNAs.
Equipment
- Personal computer (an internet connection is needed) (Intel Core i5-2300 CPU, 8 GB RAM)
Sequence data and software
- Sequence data compilation
The coding sequence (CDS) and protein sequences of interested genomes should be downloaded from relevant databases. A recommended database containing a relatively large number of sequenced plant genomes is Phytozome (http://www.phytozome.org/) (Goodstein et al., 2012). MicroRNAs of interested genomes can be retrieved from the miRBase (http://www.mirbase.org/) (Kozomara and Griffiths-Jones, 2014) or from in-house sequenced data. - Required software and online tools
The following software should be locally installed in your computer:- Hmmer 3.0 (http://hmmer.janelia.org/) (Johnson et al., 2010), for perform local hidden Markov models (HMM) search of NBS homologous proteins.
- NCBI BLAST+ or NCBI BLAST (http://blast.ncbi.nlm.nih.gov/Blast.cgi), for perform local blastp search of NBS homologous proteins.
- ActivePerl 5.14.2 (http://www.activestate.com/activeperl/downloads), for running scripts written in perl language.
- The online tools to be used are:
- COILS (http://www.ch.embnet.org/software/COILS_form.html) (Lupas et al., 1991), a program for identification of coiled-coils (CC) domain in protein sequences.
- Pfam (http://pfam. sanger.ac.uk/) (Finn et al., 2014), a database for identification of protein domains.
- psRNATarget (http://plantgrn.noble.org/psRNATarget/) (Dai and Zhao, 2011), a website designed for prediction microRNA targets in plants.
- Hmmer 3.0 (http://hmmer.janelia.org/) (Johnson et al., 2010), for perform local hidden Markov models (HMM) search of NBS homologous proteins.
Procedure
- Preparation of the query file and local database
For a given plant genome of interest, do the following:- Download all CDS and protein sequences of all protein-coding genes from a relevant database such as phytozome (http://www.phytozome.org/) (Goodstein et al., 2012). Make sure that each gene has the same name in both CDS and protein sequence files.
- Download the HMM profile and the extended amino acid sequence for NB-ARC domain (Pfam no. PF00931) from the Pfam database (http://pfam.sanger.ac.uk/) (Finn et al., 2014).
- Download all microRNA sequences from a relevant database such as miRBase (http://www.mirbase.org/) (Kozomara and Griffiths-Jones, 2014) or prepare a fasta file including all the microRNA sequences obtained in house experiments.
- Create a local database of your downloaded protein sequences for blast search using the formatdb program of the blast software (http://blast.ncbi.nlm.nih.gov/Blast.cgi).
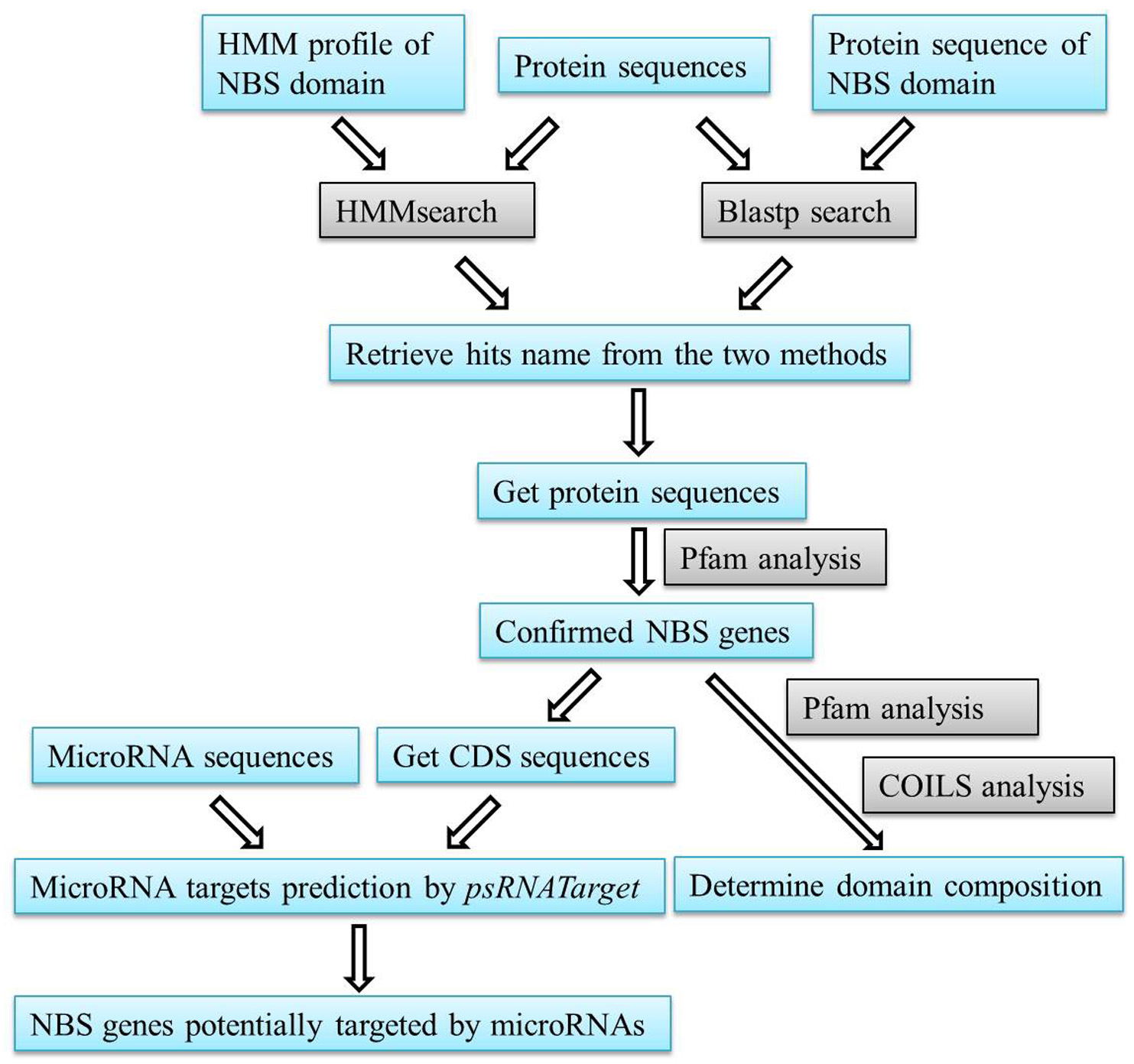
Figure 1. A flow chart of the steps described in our procedure
- Download all CDS and protein sequences of all protein-coding genes from a relevant database such as phytozome (http://www.phytozome.org/) (Goodstein et al., 2012). Make sure that each gene has the same name in both CDS and protein sequence files.
- Computational identification of NBS genes
- Perform the HMM search against the fasta file that contains protein sequences you downloaded using the hmmsearch.exe program in the hmmer 3.0 package (Johnson et al., 2010) with the HMM profile of NB-ARC domain as a query in default settings.
- Run a local BLASTp search against the protein database that was created in the procedure step A4 using the amino acid sequence of the NB-ARC domain as a query. The threshold expectation value was set to 1.0 as used in previous studies (Li et al., 2010; Shao et al., 2014).
- Retrieve the gene name of potential NBS genes from the results of HMM search and BLAST search and combined them together to obtain the maximal number of hits.
- These gene names are used to retrieve the protein sequences from protein dataset downloaded in procedure step A1. This step could be achieved manually if only a few NBS genes are found in the genome. For large datasets, we recommend the researchers writing a Perl script for this step (a script is also available from the authors upon request).
- The obtained sequences are further subjected to the online Pfam analysis to verify whether they indeed possess the NBS domain, with the E-value setting to 10-4 (Figure 2A). Sequences that do not have a detectable NB-ARC domain are discarded. The remaining sequences represent all NBS proteins of the dataset.
- The Pfam analysis is also important to detect whether these proteins have an N-terminal toll/interleukin-1 receptor (TIR) domain or RESISTANCE TO POWDERY MILDEW8 (RPW8) domain or a C-terminal LRR domain (Meyers et al., 2003). Protein sequences that do not have a detectable N-terminal domain by Pfam are further analyzed using the COILS Server (http://www.ch.embnet.org/software/COILS_form.html) (Lupas et al., 1991) in default settings to detect whether they have a coiled-coils domain at the N-terminal.
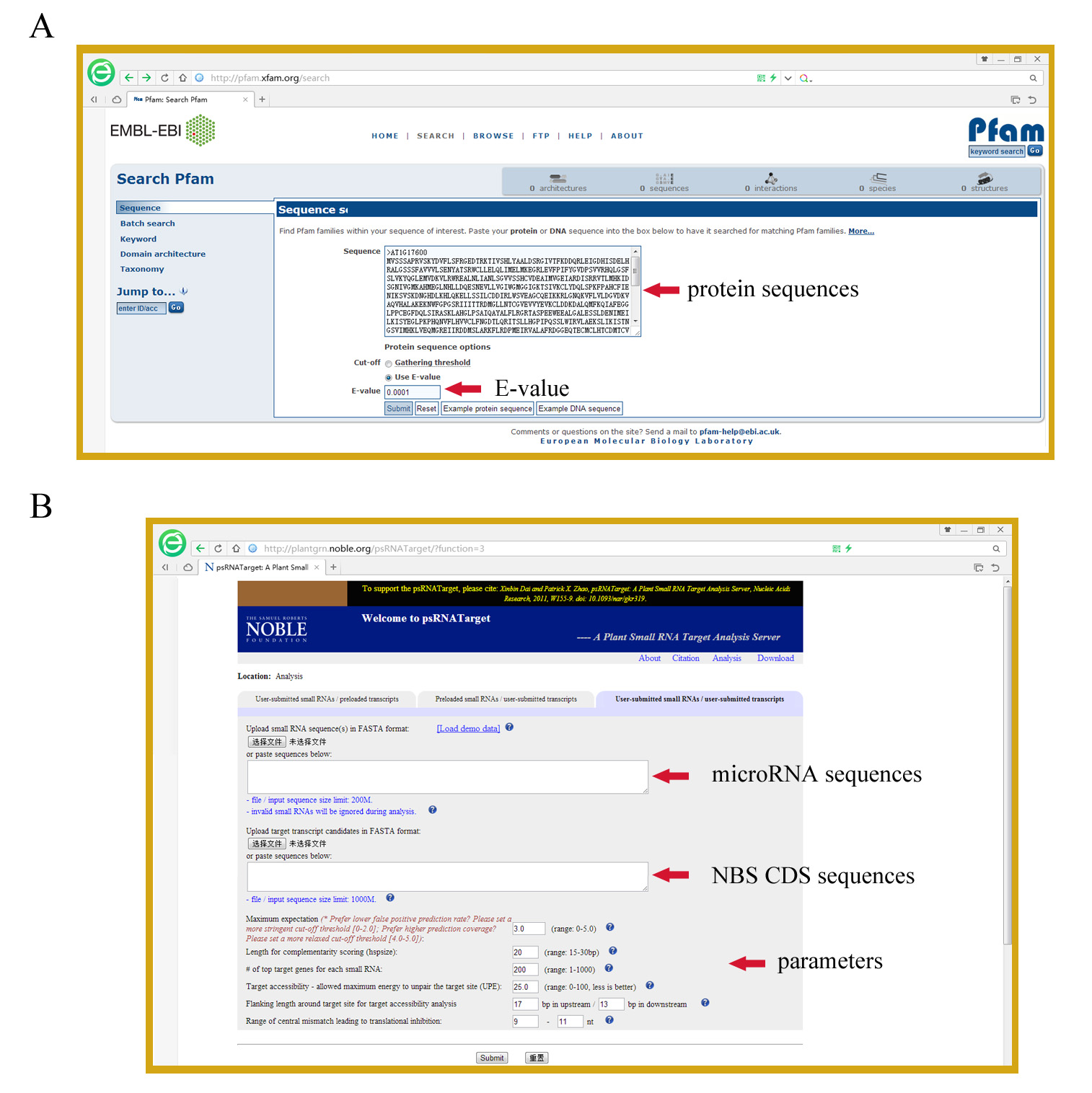
Figure 2. Screen shots for steps A) B5, and B) C2
- Perform the HMM search against the fasta file that contains protein sequences you downloaded using the hmmsearch.exe program in the hmmer 3.0 package (Johnson et al., 2010) with the HMM profile of NB-ARC domain as a query in default settings.
- Identification of NBS genes potentially targeted by microRNAs
- To predict NBS genes targeted by microRNAs, retrieve the CDS sequences of identified NBS genes by searching gene names from the downloaded CDS dataset.
- Submit the sequences corresponding to CDS of identified NBS genes and to microRNAs to the psRNATarget Server (http://plantgrn.noble.org/psRNATarget/) (Dai and Zhao, 2011) in fasta format for microRNA target prediction (Figure 2B).
Note: At this step, researchers can change the parameter settings to restrict or expand the number of predicted targets. For example, one can set the Maximum expectation (transformed from mismatch penalty) to 3 to obtain fewer hits with lower false positive prediction rate; or set the Maximum expectation to 5 to maximize the number of potential targets at a higher risk of false positive prediction rate. - Download the prediction results from the psRNATarget Server and retrieve those NBS genes predicted to be targeted by microRNAs.
- The predicted regulation of NBS genes by microRNAs could be experimentally validated by co-expression of them in tobacco leaves as described in several studies (Liu et al., 2014; Yu and Pilot, 2014).
- To predict NBS genes targeted by microRNAs, retrieve the CDS sequences of identified NBS genes by searching gene names from the downloaded CDS dataset.
Acknowledgments
This protocol is adapted from Shao et al. (2014). This work was supported by the National Natural Science Foundation of China (30930008, 31170210, 91231102 and 31400201), China Postdoctoral Science Foundation (2013M540435 and 2014T70503).
References
- Dai, X. and Zhao, P. X. (2011). psRNATarget: a plant small RNA target analysis server. Nucleic Acids Res 39(Web Server issue): W155-159.
- Finn, R. D., Bateman, A., Clements, J., Coggill, P., Eberhardt, R. Y., Eddy, S. R., Heger, A., Hetherington, K., Holm, L., Mistry, J., Sonnhammer, E. L., Tate, J. and Punta, M. (2014). Pfam: the protein families database. Nucleic Acids Res 42(Database issue): D222-230.
- Goodstein, D. M., Shu, S., Howson, R., Neupane, R., Hayes, R. D., Fazo, J., Mitros, T., Dirks, W., Hellsten, U., Putnam, N. and Rokhsar, D. S. (2012). Phytozome: a comparative platform for green plant genomics. Nucleic Acids Res 40(Database issue): D1178-1186.
- Johnson, L. S., Eddy, S. R. and Portugaly, E. (2010). Hidden Markov model speed heuristic and iterative HMM search procedure. BMC Bioinformatics 11: 431.
- Kozomara, A. and Griffiths-Jones, S. (2014). miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res 42(Database issue): D68-73.
- Li, J., Ding, J., Zhang, W., Zhang, Y., Tang, P., Chen, J. Q., Tian, D. and Yang, S. (2010). Unique evolutionary pattern of numbers of gramineous NBS-LRR genes. Mol Genet Genomics 283(5): 427-438.
- Liu, Q., Wang, F. and Axtell, M. J. (2014). Analysis of complementarity requirements for plant microRNA targeting using a Nicotiana benthamiana quantitative transient assay. Plant Cell 26(2): 741-753.
- Lupas, A., Van Dyke, M. and Stock, J. (1991). Predicting coiled coils from protein sequences. Science 252(5009): 1162-1164.
- Meyers, B. C., Kozik, A., Griego, A., Kuang, H. and Michelmore, R. W. (2003). Genome-wide analysis of NBS-LRR-encoding genes in Arabidopsis. Plant Cell 15(4): 809-834.
- Shao, Z. Q., Zhang, Y. M., Hang, Y. Y., Xue, J. Y., Zhou, G. C., Wu, P., Wu, X. Y., Wu, X. Z., Wang, Q., Wang, B. and Chen, J. Q. (2014). Long-term evolution of nucleotide-binding site-leucine-rich repeat genes: understanding gained from and beyond the legume family. Plant Physiol 166(1): 217-234.
- Shivaprasad, P. V., Chen, H. M., Patel, K., Bond, D. M., Santos, B. A. and Baulcombe, D. C. (2012). A microRNA superfamily regulates nucleotide binding site-leucine-rich repeats and other mRNAs. Plant Cell 24(3): 859-874.
- Yu, S. and Pilot, G. (2014). Testing the efficiency of plant artificial microRNAs by transient expression in Nicotiana benthamiana reveals additional action at the translational level. Front Plant Sci 5: 622.
- Zhai, J., Jeong, D. H., De Paoli, E., Park, S., Rosen, B. D., Li, Y., Gonzalez, A. J., Yan, Z., Kitto, S. L., Grusak, M. A., Jackson, S. A., Stacey, G., Cook, D. R., Green, P. J., Sherrier, D. J. and Meyers, B. C. (2011). MicroRNAs as master regulators of the plant NB-LRR defense gene family via the production of phased, trans-acting siRNAs. Genes Dev 25(23): 2540-2553.
- Zhang, Y. M., Shao, Z. Q., Wang, Q., Hang, Y. Y., Xue, J. Y., Wang, B. and Chen, J. Q. (2015). Uncovering the dynamic evolution of nucleotide-binding site-leucine-rich repeat (NBS-LRR) genes in Brassicaceae. J Integr Plant Biol. (Epub ahead of print)
Article Information
Copyright
© 2015 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Shao, Z., Zhang, Y., Wang, B. and Chen, J. (2015). Computational Identification of MicroRNA-targeted Nucleotide-binding Site-leucine-rich Repeat Genes in Plants. Bio-protocol 5(21): e1637. DOI: 10.21769/BioProtoc.1637.
-
Shao, Z. Q., Zhang, Y. M., Hang, Y. Y., Xue, J. Y., Zhou, G. C., Wu, P., Wu, X. Y., Wu, X. Z., Wang, Q., Wang, B. and Chen, J. Q. (2014). Long-term evolution of nucleotide-binding site-leucine-rich repeat genes: understanding gained from and beyond the legume family. Plant Physiol 166(1): 217-234.
Category
Plant Science > Plant molecular biology > RNA
Systems Biology > Interactome > Gene network
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.