- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Telomere-mediated Chromosomal Truncation via Agrobacterium tumefaciens or Particle Bombardment to Produce Engineered Minichromosomes in Plants
(*contributed equally to this work) Published: Vol 5, Iss 18, Sep 20, 2015 DOI: 10.21769/BioProtoc.1595 Views: 9257
Reviewed by: Fanglian HeAnonymous reviewer(s)
Original research article
The authors used this protocol in:

May 2008
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
Minichromosomes are small, autonomously functioning chromosomes that exist separately from the normal chromosomal set. Creation of minichromosomes in plants relies on telomere truncation to remove the chromosome arms and the native telomere sequence and replace them with a transgene together with a new telomere sequence to generate a modifiable small chromosome. Telomere truncation has been accomplished utilizing both Agrobacterium tumefaciens, in which a telomere repeat sequence is cloned into the transformation vector near the right border, and particle bombardment, in which the genes of interest and telomere sequence are co-introduced into the plant. In this protocol we will describe the methods for introducing telomere sequences to both Agrobacterium and gold particles, as well as the methods required to verify that these sequences are intact.
[Introduction] Engineered minichromosomes are autonomously functioning chromosomes that contain all of the necessary components required for maintenance through the cell cycle. The production of engineered minichromosomes has several potential applications for the next generation of genetic engineering (Gaeta et al., 2012). The construction of such chromosomes by assembling a centromere, origin of replication, and selectable marker all capped by telomere sequences, as originally performed in yeast, is not feasible in plants because of the epigenetic nature of centromere sequences (Birchler and Han, 2009; Birchler et al., 2011; Liu et al., 2015). In other words, functional centromeres in plants are determined by chromatin features independent of the underlying DNA and therefore the cloning and re-introduction of centromere sequences will not produce a minichromosome. In contrast to the centromere, the telomere is reliant on sequence, with most plant telomeres containing the same TTTAGGG repeat (Adams et al., 2001). As a result, introduction of telomere sequences during transformation has the potential to confer telomere function.
Because of the epigenetic state of the centromere, engineered minichromosomes in plants need to be produced by cleaving away the chromosome arms from an endogenous centromere that never leaves a cell, a procedure known as the top-down method. This was accomplished with the finding that introduction of the chromosome end sequences, the telomere, would cleave chromosomes at the site of integration (Yu et al., 2006). By including genes of interest in addition to the telomere sequences, the foundation to build engineered minichromosomes to specification was established. This protocol describes the procedure to generate these initial truncated minichromosomes.
The process of creating minichromosomes utilizes standard plant transformation protocols. The only modification is the addition of telomere sequences to the transformation construction so that both a transgene and telomere sequence are introduced into a double stranded break during the transformation process. In some cases, the introduced telomere sequence is recognized by telomere elongation machinery and converted into a functioning telomere. As a result, the acentric fragment distal to the insertion point will be lost, and a minichromosome will be created.
Telomere truncation works well with both Agrobacterium tumefaciens and particle bombardment transformation techniques. Using Agrobacterium, successful minichromosome creation relies on inclusion of telomere sequences in the transformation construct. With particle bombardment, telomere sequences are simply added to the DNA mixture that is adhered to the gold beads before transformation is performed. While the length of telomere sequence required for telomere truncation is not known, a study in Arabidopsis thaliana successfully created truncated chromosomes with telomere repeats as short as 100 bp (Nelson et al., 2011). The study also found, however, that longer telomere sequences were more likely to induce truncation events. As a result, it is suggested that the largest amount of telomere that can be reasonably obtained be used during transformation.
While the concept of including telomere sequences in transformation is relatively simple, working with telomere sequences using current molecular cloning techniques is challenging. The repeated nature of the sequences and the high GC content are inhibitive to polymerase function. As a result, protocols reliant on polymerase function, such as PCR or Sanger sequencing, are not efficient. Additionally, oligonucleotide synthesis technologies are limited for producing telomere repeat sequences at the time of this writing. Traditional cloning utilizing restriction enzymes has been most successful in our work. Isolated telomere sequences, when subjected to agarose gels, do not migrate at the expected sizes, but instead are found as discreet bands or smears throughout the gel, probably because they adopt various secondary structures. Additionally, purification of telomere sequences with gel or column purification is usually inefficient unless the DNA is present in large amounts, making traditional cloning difficult. Adding to these challenges is the observation that long telomere sequences are unstable in microbial cells, and have a tendency to be deleted and shortened over time. As a result, Stbl cells (Invitrogen), which possess the recA1 genotype and are specifically designed to prevent repeated sequences from recombination and thus rearrangement, must be used to maintain the repeat, and multiple clones should be isolated and screened to ensure the full size is present. Additionally, when a clone has been isolated, which contains the desired telomere insert, it is often useful to make a large plasmid extraction that is stored in addition to bacterial stocks.
In order to generate engineered minichromosomes, the protocols presented below were developed. For cloning purposes, the telomere sequence is excised from a gel and ligated to the target plasmid within the agarose mixture. The source of the telomere sequence is plasmid pWY82 (Yu et al., 2006), which contains 2.6 kb of the telomere repeat (TTTAGGG). For particle bombardment, primers are used with a modified PCR program to amplify the telomere repeats, which are gel purified and added to gold particles together with the construct of interest. Whether truncation will be performed with Agrobacterium or particle bombardment, the standard transformation protocol for the species of interest can be followed. Fluorescence in-situ hybridization is then performed to determine if a minichromosome has successfully been produced (Yu et al., 2007).
Materials and Reagents
- Target binary plasmid with compatible restriction enzyme cut sites near right border or purified plasmid for co-bombardment
Note: There are many binary vectors available, and any are acceptable for use provided the vector contains the necessary restriction enzyme cut sites to move the telomere fragment from pWY82. A map and sequence of pWY82 can be obtained by contacting the corresponding author. In addition, it is suggested to place the telomere sequences near the right border as in the original truncation plasmid (Yu et al., 2007). It is not known whether placing it inside the left border is effective. - Plasmid pWY82 (Contact corresponding author)
- QIAprep Spin Miniprep Kit (QIAGEN, catalog number: 27104 )
- AmbionTM Nuclease-Free Water (not DEPC-Treated) (Fisher Scientific, catalog number: AM9937 )
- Restriction Enzymes (New England Biolabs)
Note: Enzymes must be chosen based on compatibility between pWY82 and target vector. - UltraPureTM Low Melting Point Agarose (Life Technologies, InvitrogenTM, catalog number: 16520-050 )
- Trizma® base (Sigma-Aldrich, catalog number: T1503 )
- Ethylenediaminetetraacetic acid (EDTA) (Sigma-Aldrich, catalog number: E6758 )
- Acetic Acid (Sigma-Aldrich, catalog number: 27225 )
- Antarctic Phosphatase (New England BioLabs, catalog number: M0289S )
- DNA Gel Loading Dye (6x) (Thermo Fisher Scientific, catalog number: R0611 )
- GeneRuler 1 kb DNA Ladder (Thermo Fisher Scientific, catalog number: SM0311 )
- Ethidium Bromide (Sigma-Aldrich, catalog number: E7637 )
- T4 DNA Ligase (New England BioLabs, catalog number: M0202S )
- ElectroMAX Stbl4 Cells (、Thermo Fisher Scientific, catalog number: 11635-018 )
- S.O.C Media (Super Optimal Broth with Catabolic repressor) (Thermo Fisher Scientific, catalog number: 15544-034 )
- Agar (Sigma-Aldrich, catalog number: A1296 )
- Petri Dishes
- LongAmp® Taq DNA Polymerase (New England BioLabs, catalog number: M0323S )
- Fisher BioReagents LB Broth, Miller (Granulated) (Fisher Scientific, catalog number: BP9723-2 )
- Spectinomycin dihydrochloride pentahydrate (Sigma-Aldrich, catalog number: S4014 )
- 2x YT medium (Sigma-Aldrich, catalog number: Y2377 )
- LB broth (see Recipes)
- LB plates (see Recipes)
- 2x YT broth (see Recipes)
- Spectinomycin (see Recipes)
- TAE (see Recipes)
Equipment
- 30 °C incubator
- 30 °C shaker
- 4 °C cold room
- 250 ml baffled culture flasks
- Nanodrop spectrophotometer (Thermo Fisher Scientific)
- Vacuum concentrator
- Gel electrophoresis system
- 37 °C waterbath
- 70 °C waterbath
- Ultraviolet transilluminator
- Electroporator
- Thermalcycler
- Wizard® SV Gel and PCR Clean-Up System (Promega Corporation, catalog number: A9281 )
Procedure
- In-Gel ligation
- Streak a LB plate containing 100 mg/ml spectinomycin with plasmid pWY82 and grow at 30 °C for 2 days.
- Begin a 3 ml starter culture with a single colony of pWY82 in 2XYT media with 100 mg/ml spectinomycin and grow for 48 h at 30 °C and 250 rpm.
Note: pWY82 is a slow growing plasmid. - Add 500 μl of starter culture to 125 ml 2XYT in a baffled culture flask. Shake at 250 rpm for 48 h at 30 °C until OD600 = ~2 when measured with a spectrophotometer.
- Extract plasmid DNA with the QIAprep Spin Miniprep kit, using 4 ml
of culture per spin column. Elute with 50 μl of nuclease free water preheated
to 50 °C.
- Pool minipreps into a 1.7 ml tube and place into a vacuum concentrator until
the concentration is about 1 μg/μl when measured with a Nanodrop spectrophotometer.
- Confirm the integrity of pWY82, target plasmid, and cut sites by digesting 1 μg of pWY82 and target plasmid with each enzyme individually and in tandem following the manufacturer’s instructions.
Note: Due to the instability of telomere repeats, the telomere fragment will show multiple bands when subjected to agarose gel electrophoresis with the full 2.6 kb repeat not always visible (Figure 1).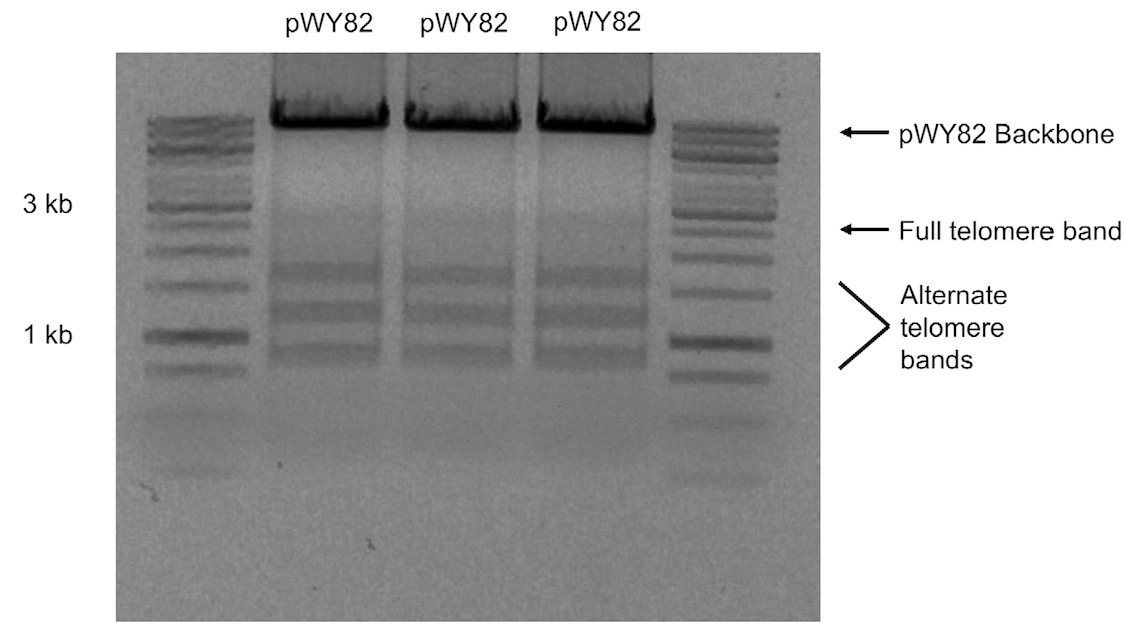
Figure 1. 1% (w/v) agarose gel of three different colonies of pWY82 cut with EcoRI and HindIII showing the range of telomere sizes - Digest 10 μg of pWY82 and 5 μg of target plasmid with 10 units of each restriction enzyme for at least one hour.
Note: Total restriction digest volume should be no more than 50 μl. - Pour a 1% (w/v) low melting point agarose gel prepared with 1x TAE in a 4 °C cold room.
Note: Use a comb large enough to fit each restriction digest fully in a single well. - Treat target plasmid with 5 units of Antarctic phosphatase for 15 min at
37 °C.
- Add 15 μl of 6x loading dye to each reaction.
- Fill the electrophoresis chamber with cold 1x TAE and carefully insert the
gel.
Caution: Low melting point gels are extremely fragile. - Load each restriction digest completely into the gel.
- Load 6 μl of GeneRuler 1 kb DNA Ladder next to each restriction digest lane.
- Run the gel in a 4 °C cold room at 100 V until the lower band of loading dye
has reached the bottom of the gel.
- Carefully transfer the gel to a dish containing 1x TAE and 0.5 μg /ml ethidium bromide and stain for 30 min.
- Visualize DNA with UV light and estimate concentrations by comparison to the reference DNA ladder.
Note: Concentration is estimated by comparing sample UV intensity with ladder UV intensity. The amount of DNA present in the ladder is available in the literature. - Carefully excise the uppermost visible telomere band and the target plasmid backbone with a scalpel and place into separate 1.7 ml tubes (Figure 2).
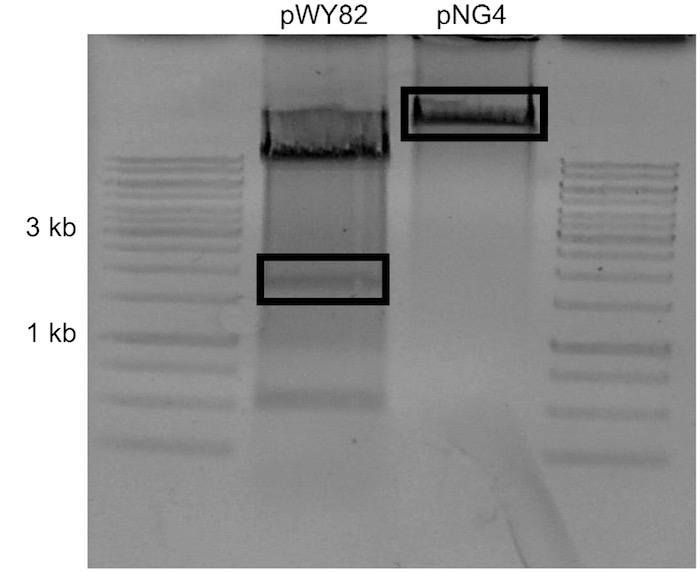
Figure 2. Example of gel to be used for ligation. pNG4 was the target plasmid for this ligation. The boxed sections were excised. - Add 1 ml of nuclease free water to gel slices and allow to dialyze at 4 °C overnight to remove salts from agarose.
- Remove water from tubes and place in a 70 °C water bath until completely melted (about 5 min).
Note: Flick tubes each minute to ensure agarose melts completely. - Once melted, move tubes to 37 °C water bath.
- Prepare ligase mixture in a separate 1.7 ml tube while agarose cools (Table 1).
Table 1. Ligation reaction componentsComponent Amount Ligase buffer 5 μl T4 DNA ligase 1 μl (400 units) Target plasmid backbone 100 ng as estimated from gel Telomere To 50 μl - After cooling about 5 min, add target plasmid and telomere to ligation
mixture.
Note: Work quickly so agarose does not solidify in pipette tip. - Mix by gentle pipetting and allow to return to room temperature.
Note: The reaction should solidify as it cools. - Incubate at room temperature overnight.
- Dialyze reaction by adding 1 ml of water to tube and incubate at room
temperature for 15 min.
Note: Flick tube until agarose is freely floating in water. - Begin to thaw Stbl4 cells on ice.
Note: 40 μl of cells are needed per reaction. - Completely remove water from dialysis and add 50 μl of fresh nuclease free
water. Place tube in a 70 °C water bath for 10 min, or until completely melted.
Note: Gently flick tube every minute to ensure gel is completely melted. - Add 2 μl of ligation reaction to 40 μl of Stbl4 cells and electroporate following
manufacturers recommended settings for the electroporator. Resuspend electroporated cells in 500 ml of S.O.C media and shake in 15 ml culture tubes for 1.5 h at 250 rpm at 30 °C.
- Plate 100 μl of the transformation preparation onto pre-warmed LB plates containing the appropriate antibiotic and incubate at 30 °C until colonies appear.
Note: Due to the lower temperature, colonies may take 2 days to appear on plates. - Screen for positive colonies with either colony PCR or colony hybridization.
Note: Screening for positive colonies:
Due to the repetitive nature of the telomere, it is not advisable to screen colonies by amplifying across the telomere insertion site, as most PCR reactions will fail. Instead, colony PCR can be carried out using a forward primer that anneals 5’ to the insertion site in the target plasmid, and a reverse primer that anneals to a site 5’ to the telomere repeat in pWY82 that is also inserted into the target plasmid as shown in Figure 3 below. It is important to ensure good primer function in each plasmid before attempting colony PCR. The primer pWY82R with the sequence 5’ GTGGGACCCGAGGGAATC 3’ has been routinely used as the reverse primer in colony PCR.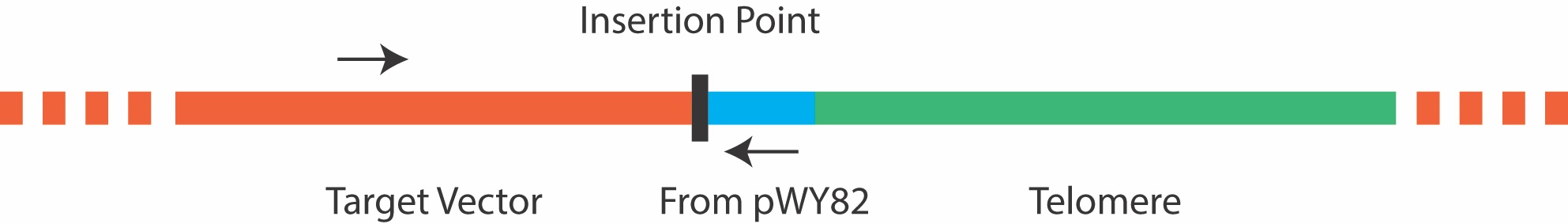
Figure 3. Primers for colony PCR should be chosen that border the insertion point, and do not amplify the telomere repeat
Alternatively, entire plates of colonies can be screened for the presence of telomere by colony hybridization (Sambrook et al., 1989). This method allows for rapid screening of hundreds of colonies at once, and is extremely sensitive. The oligonucleotide probe used for the hybridization consists of 10 units of the telomere repeat, either in the 5’ or 3’ direction. Finally, it is suggested to sequence into the telomere repeat to ensure that the insert is in the correct orientation.
- Streak a LB plate containing 100 mg/ml spectinomycin with plasmid pWY82 and grow at 30 °C for 2 days.
- Detecting telomere insert size
Once positive colonies have been obtained, it is important to screen colonies for telomere size. For most purposes, the colony with the largest telomere insert possible is desired for transformation. Telomere size estimation is best carried out through Southern transfer and hybridization (Southern, 1975). The target plasmid, as well as plasmid pWY82, is run alongside the tested colonies to serve as controls.- Inoculate positive colonies, plus one culture of pWY82 and one culture of original target plasmid, into 3 ml of 2XYT media containing the appropriate antibiotics. Shake culture at 250 rpm at 30 °C for 8 h.
- Inoculate a fresh 5 ml 2XYT culture with 100 μl of starter culture of each colony containing the appropriate antibiotics and shake at 250 rpm for 16-18 h at 30 °C.
Note: It is sometimes useful to inoculate and grow larger (25 ml) cultures of colonies that have been chosen. An aliquot can be used for screening, and when a clone has been identified with the desired telomere fragment, the remaining culture can then be used for a larger scale plasmid preparation. - Using 4 ml of each culture, extract plasmid DNA with the QIAprep Spin Miniprep Kit. Elute plasmid extraction into 40 μl of warm (55 °C) nuclease free water.
- Perform a restriction digest on 1 μg of each plasmid in a 20 μl total volume using enzymes that will cut as close to the telomere insert as possible on both ends so the telomere insert is excised. Additionally, cut pWY82 with enzymes that will also excise telomere insert for reference.
- Pour a standard 1% (w/v) 1x TAE agarose gel using combs large enough to fit the entire restriction digest.
- Once the restriction digest is complete, add 5 μl 6x loading dye to each reaction.
- Load each digest into the agarose gel and run at 100 V until the lower band has reached the bottom of the gel.
- Carefully transfer the gel to a dish containing 1x TAE and 0.5 μg/ml ethidium bromide and stain for 30 min.
- Visualize DNA with UV light.
Note: Due to the secondary structure of telomere, estimation of telomere size from a restriction digest will be unreliable. - Transfer the DNA to a nitrocellulose membrane by Southern transfer (Green and Sambrook, 2012).
- Hybridize the membrane with a radiolabeled oligonucleotide and compare to the size standard to estimate telomere size (Green and Sambrook, 2012).
Note: An oligonucleotide of sequence (TTTAGGG)10 is extremely sensitive for detecting the presence of telomere.
- Inoculate positive colonies, plus one culture of pWY82 and one culture of original target plasmid, into 3 ml of 2XYT media containing the appropriate antibiotics. Shake culture at 250 rpm at 30 °C for 8 h.
- Telomere PCR protocol to generate telomere fragments
Telomere fragments of varying sizes can be generated using PCR for use in the co-bombardment method of minichromosome production. With the primers noted below, and running the reaction as shown, will generate a variety of telomere conglomerates of differing sizes can be produced by cross annealing of the primers. These telomere lengths can be visualized by gel electrophoresis. Telomere DNA of a particular length can be obtained by cutting out the size of telomere desired from the agarose gel. These steps below will outline how to generate telomere fragments via PCR and how to utilize the DNA obtained. This protocol is adapted from a similar protocol used to make fluorescent probes that label telomere (IJdo et al., 1991).- Develop PCR primers
- Use the sequences below for primer development. These will allow for the production of large telomere fragments.
- Telomere primer sense: 5’ (TTTAGGG)10 3’.
- Telomere primer antisense: 5’ (CCCTAAA)10 3’
- Primers can be diluted to the micromolar concentrations listed on the table below which result in differing telomere fragment sizes.
1.25 micromoles of Forward Telomere Primer
1.25 micromoles of Reverse Telomere PrimerProduces Larger Fragments (see Figure 4) 25 micromoles of Forward Telomere Primer
25 micromoles of Reverse Telomere PrimerProduces Shorter Fragments (see Figure 4) - Use the sequences below for primer development. These will allow for the production of large telomere fragments.
- PCR reaction assembly
- Use a proofreading Taq polymerase, such as LongAmp Taq DNA Polymerase from New England Biolabs, and use the above concentration of primers. At least four reactions are recommended in order to obtain enough DNA for use after DNA gel extraction. PCR reaction assembly is as follows:
PCR Reaction Assembly using LongAmp Taq Polymerase from NEB
Nuclease Free Water 12 microliters 24 microliters 5x LongAmp Taq Buffer 4 microliters 8 microliters Sense Telomere Primer 0.5 microliters 1 microliter Antisense Telomere Primer 0.5 microliters 1 microliter 10mM dNTPs 2 microliters 4 microliters LongAmp Taq Polymerase 1 microliters 2 microliters 20 microliters per reaction 40 microliters per reaction
Thermocycler protocol98 °C 10 sec
55 °C 20 sec
72 °C 5 secRepeat 5x 98 °C 20 sec
55 °C 20 sec
72 °C 5 secRepeat 5x 98 °C 20 sec
55 °C 20 sec
72 °C 10 secRepeat 5x 98 °C 20 sec
62 °C 20 sec
72 °C 30 secRepeat 5x 72 °C 5 min 4 °C ∞
- Use a proofreading Taq polymerase, such as LongAmp Taq DNA Polymerase from New England Biolabs, and use the above concentration of primers. At least four reactions are recommended in order to obtain enough DNA for use after DNA gel extraction. PCR reaction assembly is as follows:
- Use gel electrophoresis to determine the size of fragments generated (Figure 4).
Notes:- The smears of stained DNA on the gel image in Figure 4 are telomere fragments of varying size. Figure 4 is the result of 40 microliter PCR reactions. Cutting out the smear associated with the desired size indicated by the ladder provides telomere fragments of that length.
- The smears of telomere fragments in Figure 4 vary in size based on the Taq polymerase used as well as the primer concentration.
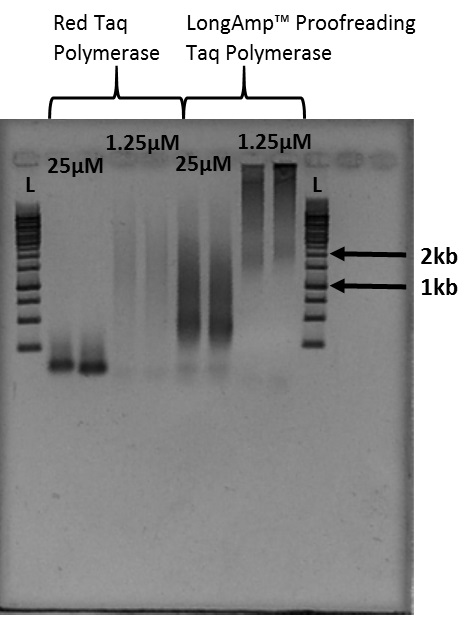
Figure 4. Example of Telomere PCR product imaged after gel electrophoresis: L indicates the 1kb ladder used for this experiment. 1.25 micromoles and 25 micromoles indicate the concentration of PCR primer present in each sample. - Follow the gel electrophoresis protocol (Sambrook and Russell, 2001).
- The smears of stained DNA on the gel image in Figure 4 are telomere fragments of varying size. Figure 4 is the result of 40 microliter PCR reactions. Cutting out the smear associated with the desired size indicated by the ladder provides telomere fragments of that length.
- Gel extraction of telomere DNA
- Excise the piece of gel corresponding to the size of telomere desired, and extract the telomere DNA from this gel piece. For example, if a 2 kb telomere fragment is desired, cut out the gel smear closest in relation to the 2 kb mark on the ladder. Generally, larger fragments of telomere are desired if the goal is truncation of chromosomes because the larger the telomere fragment, the better chance that an insertion of the telomere fragment will be recognized by the telomere capping machinery as the new end for the chromosome. The minimum size of telomere fragment required for this recognition may vary from species to species (Teo et al., 2011).
- Use the Wizard SV Gel and PCR Clean-Up System for extracting DNA from agarose gels. Follow the manufacturer’s instructions for extracting DNA from the agarose gel piece.
Notes:- A significant amount of DNA will be lost when performing gel extraction. It is recommended to use a large volume PCR reaction or multiple reactions.
- It is recommended that elutions are performed using nuclease-free water, such as Nuclease-Free Water from Life Technologies, to allow for further use in cloning if desired.
- A significant amount of DNA will be lost when performing gel extraction. It is recommended to use a large volume PCR reaction or multiple reactions.
- Excise the piece of gel corresponding to the size of telomere desired, and extract the telomere DNA from this gel piece. For example, if a 2 kb telomere fragment is desired, cut out the gel smear closest in relation to the 2 kb mark on the ladder. Generally, larger fragments of telomere are desired if the goal is truncation of chromosomes because the larger the telomere fragment, the better chance that an insertion of the telomere fragment will be recognized by the telomere capping machinery as the new end for the chromosome. The minimum size of telomere fragment required for this recognition may vary from species to species (Teo et al., 2011).
- Using the extracted DNA
- Telomere DNA fragments obtained can then be used in a cobombardment with a transgene.
- Preparation of telomere DNA as described in (Kikkert et al., 2005) can be used in a co-bombardment.
- Telomere DNA fragments obtained can then be used in a cobombardment with a transgene.
- Develop PCR primers
- Discussion
Conceptually, minichromosomes can be created by either a bottom-up or a top-down strategy. A bottom-up strategy would require the artificial creation of centromere, telomere, and origin of replication sequences to create a viable chromosome. Currently, origins of replication and centromeres cannot be made artificially in most eukaryotes. The top-down approach utilizes an existing chromosome to create a minichromosome by truncating it with a telomere-containing construct. Telomere-mediated truncation is an effective means of creating top-down minichromosomes. The essential factor for telomere-mediated truncation, the telomere sequences, can be difficult to manipulate because of their constitution as a large array of repeats. The methods for creating and utilizing telomere DNA described here are the most effective. After cloning telomere into a vector, or preparing telomere for cobombardment, proceed normally with transformation protocols. Tissue regeneration is the same as for standard transformation. Selection for truncation events can be performed using cytological techniques, such as fluorescent in situ hybridization (Yu et al., 2007). These methodologies should allow for the creation of telomere DNA of desired size, inclusion into a vector if so desired, introduced into the species of choice, and selection for truncated chromosomes.
Recipes
- LB broth
For 500 ml of culture media, dissolve 12.5 g of LB media in 400 ml of water
Bring final volume to 500 ml and autoclave for 20 min - LB plates
For 500 ml of media, dissolve 12.5 g of LB media and 6 g of agar in 400 ml of water
Bring final volume to 500 ml and autoclave for 20 min
Allow to cool to 50 °C and add appropriate antibiotics
Gently swirl and add thin layer to petri dishes
Allow to cool then invert and store at 4 °C - 2x YT broth
For 500 ml of culture media dissolve 15.5 g of 2x YT powder in 400 ml of water
Bring final volume to 500 ml and autoclave for 20 min - Spectinomycin (100 mg/ml)
For 10 ml of stock, add 1 g of spectinomycin to 10 ml of autoclaved water in a 15 ml conical tube
Invert until completely dissolved and filter sterilize with a 0.2 micron filter
Aliquot 500 μl into 1.7 ml tubes and store at -20 °C - TAE
To make 1 L of 50x TAE stock add 242 g trizma base, 14.6 g ethylenediaminetetraacetic acid (EDTA), and 57.1 ml of acetic acid to 500 ml of water and dissolve
Bring total volume to 1 L with water
Dilute to 1x working stock with water
Acknowledgments
Research on this topic was supported by National Science Foundation grant IOS-1339198 from the Plant Genome Program.
References
- Adams, S. P., Hartman, T. P., Lim, K. Y., Chase, M. W., Bennett, M. D., Leitch, I. J. and Leitch, A. R. (2001). Loss and recovery of Arabidopsis-type telomere repeat sequences 5'-(TTTAGGG)(n)-3' in the evolution of a major radiation of flowering plants. Proc Biol Sci 268(1476): 1541-1546.
- Birchler, J. A., Gao, Z., Sharma, A., Presting, G. G. and Han, F. (2011). Epigenetic aspects of centromere function in plants. Curr Opin Plant Biol 14(2): 217-222.
- Birchler, J. A. and Han, F. (2009). Maize centromeres: structure, function, epigenetics. Annu Rev Genet 43: 287-303.
- Gaeta, R. T., Masonbrink, R. E., Krishnaswamy, L., Zhao, C. and Birchler, J. A. (2012). Synthetic chromosome platforms in plants. Annu Rev Plant Biol 63: 307-330.
- Green, M.R., and Sambrook, J. (2012). Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press.
- Ijdo, J. W., Wells, R. A., Baldini, A. and Reeders, S. T. (1991). Improved telomere detection using a telomere repeat probe (TTAGGG)n generated by PCR. Nucleic Acids Res 19(17): 4780.
- Kikkert, J. R., Vidal, J. R. and Reisch, B. I. (2005). Stable transformation of plant cells by particle bombardment/biolistics. Methods Mol Biol 286: 61-78.
- Nelson, A. D., Lamb, J. C., Kobrossly, P. S. and Shippen, D. E. (2011). Parameters affecting telomere-mediated chromosomal truncation in Arabidopsis. Plant Cell 23(6): 2263-2272.
- Sambrook, J., and Russell, D.W. (2001). Molecular cloning a laboratory manual. Cold Spring Harbor Laboratory Press.
- Sambrook, J., Fritsch, E.F., and Maniatis, T. (1989). Molecular cloning: A laboratory manual. Cold Spring Harbor Laboratory Press.
- Southern, E. M. (1975). Detection of specific sequences among DNA fragments separated by gel electrophoresis. J Mol Biol 98(3): 503-517.
- Teo, C. H., Ma, L., Kapusi, E., Hensel, G., Kumlehn, J., Schubert, I., Houben, A. and Mette, M. F. (2011). Induction of telomere-mediated chromosomal truncation and stability of truncated chromosomes in Arabidopsis thaliana. Plant J 68(1): 28-39.
- Vega, J. M., Yu, W., Han, F., Kato, A., Peters, E. M., Zhang, Z. J. and Birchler, J. A. (2008). Agrobacterium-mediated transformation of maize (Zea mays) with Cre-lox site specific recombination cassettes in BIBAC vectors. Plant Mol Biol 66(6): 587-598.
- Yu, W., Lamb, J. C., Han, F. and Birchler, J. A. (2006). Telomere-mediated chromosomal truncation in maize. Proc Natl Acad Sci U S A 103(46): 17331-17336.
- Yu, W., Han, F., Gao, Z., Vega, J. M. and Birchler, J. A. (2007). Construction and behavior of engineered minichromosomes in maize. Proc Natl Acad Sci U S A 104(21): 8924-8929.
Article Information
Copyright
© 2015 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Graham, N. D., Swyers, N. C., Gaeta, R. T., Zhao, C., Cody, J. P. and Birchler, J. A. (2015). Telomere-mediated Chromosomal Truncation via Agrobacterium tumefaciens or Particle Bombardment to Produce Engineered Minichromosomes in Plants. Bio-protocol 5(18): e1595. DOI: 10.21769/BioProtoc.1595.
Category
Plant Science > Plant molecular biology > DNA > DNA structure
Molecular Biology > DNA > DNA modification
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.