- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Complex in vivo Ligation Using Homologous Recombination and High-efficiency Plasmid Rescue from Saccharomyces cerevisiae


Published: Vol 5, Iss 13, Jul 5, 2015 DOI: 10.21769/BioProtoc.1521 Views: 14882
Reviewed by: Fanglian HeAndreea DarabaAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Sep 2011
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
The protocols presented here allow for the facile generation of a wide variety of complex multipart DNA constructs (tagged gene products, gene fusions, chimeric proteins, and other variants) using homologous recombination and in vivo ligation in budding yeast (Saccharomyces cerevisiae). This method is straightforward, efficient and cost-effective, and can be used both for vector creation and for subsequent one-step, high frequency integration into a chromosomal locus in yeast. The procedure utilizes PCR with extended oligonucleotide “tails” of homology between multiple fragments to allow for reassembly in yeast in a single transformation followed by a method for highly efficient plasmid extraction from yeast (for transformation into bacteria). The latter is an improvement on existing methods of yeast plasmid extraction, which, historically, has been a limiting step in recovery of desired constructs. We describe the utility and convenience of our techniques, and provide several examples.
[Introduction] Homologous recombination (HR) in S. cerevisiae has long been recognized as an extremely convenient method for assembling DNA fragments in vivo (Szostak et al., 1983; Ma et al., 1987; Oldenburg et al., 1997). Given the efficiency of HR in yeast, it has been exploited in ways that have both increased its utility, enhanced its versatility, and permitted its application to a broad range of experimental objectives. Improvements to this general approach include using in vivo ligation as a platform for directed mutagenesis (Muhlrad et al., 1992), introducing counterselection to aid in plasmid creation (Gunyuzlu et al., 2001; Anderson and Haj-Ahmad, 2003), and adapting in vivo assembly to vectors that cannot be propagated in yeast (Iizasa and Nagano, 2006; Joska et al., 2014). However, previous studies have not fully harnessed the power and utility of HR. We have found that HR in vivo allows for very efficient assembly and recovery of plasmids containing numerous (>5 separate pieces) fragments of DNA in a single transformation step. Thereby, we have been readily able to construct a wide variety of gene disruption cassettes and integration cassettes, to clone exceptionally large genes, to assemble multi-part chimeric genes, and to generate multiply-tagged gene products. The primary utility and power of our methods are the capability to correctly and efficiently assemble multiple DNA fragments transformed into yeast in a single step. Our approach (Figure 1A) has been used successfully: (i) to assemble in-frame chimeras between two or more different genes; (ii) to fuse gene products to fluorescent probes and/or epitope tags at either their N- and/or C-termini, or both; (iii) to create gene deletion cassettes with large amounts of untranslated flanking sequence; (iv) to introduce one or more short linker sequences or epitope tag(s) between assembled genes or gene fragments; (v) to utilize a variety of transcriptional promoters and terminators; and, importantly, (vi) in one step, to generate constructs marked with a drug resistance gene cassette or a selectable nutritional gene cassette that integrate into the genome at the desired locus. Because HR in the yeast cell carries out the in vivo construction process (and subsequent integration, if desired), no kit or proprietary system is required and the assembly of collections of plasmids can be done in a massively parallel manner. In this regard, our system is considerably less expensive than the in vitro enzyme-driven “Gibson cloning” (Gibson, 2011) procedure, yet still remarkably efficient. Also, our system (unlike those requiring restriction enzyme digests to insert gene fragments) does not result in the insertion (or loss) of any nucleotides, which can sometimes occur in classical restriction site cloning. In our method, precise control over both the coding sequence and the flanking untranslated regions (UTRs) can be achieved. Lastly, constructs generated using this system can be coupled with the haploid yeast genome deletion collection (Winzeler et al., 1999; Giaever et al., 2002) to allow for simple and efficient one-step integration at any designed locus. The only rate-limiting factor in our method is the need for the transformed yeast cells to grow for a few days before the construct (or genetically altered cell) can be recovered.
Although similar overall methods may exist (Andersen, 2011), in our protocol, we developed several important improvements, which greatly enhance efficient recovery of the DNA constructs from yeast cells, including: (i) a specific yeast genotype that is much easier to lyse than standard laboratory strains, such as S288C (and its derivatives, e.g. BY4741); (ii) a spheroplasting step (to destroy the yeast cell wall); (iii) glass bead beating for better nucleic acid extraction; and, (iv) bacteria chemically treated for ultra-efficient DNA transformation.
Materials and Reagents
- Yeast strains: SF838-1Dα (MATα ura3-52 leu2-3,122 his4-519 ade6 pep4-3 gal2; Rothman and Stevens, 1986) and THS4218 (SF838-1Dα; HIS4 his3Δ::HygR)
- KOD Hot Start DNA Polymerase (EMD Millipore, distributed by VWR International, catalog number: 80511-384 )
- DNA oligonucleotide primers (Integrated DNA Technologies, Inc.)
- DpnI restriction enzyme (New England Biolabs, catalog number: R0176S )
- Appropriate restriction enzyme(s) for digestion (New England Biolabs)
- PEG: Poly (ethylene glycol), BioXtra avg. molecular weight 3,350 (Sigma-Aldrich, catalog number: P4388-1KG )
- Lithium acetate dihydrate (Sigma-Aldrich, catalog number: L6883-1KG )
- ssDNA: Deoxyribonucleic acid sodium salt from salmon testes (Sigma-Aldrich, catalog number: D1626-1G )
- GeneJET Plasmid Miniprep Kit (Thermo Fisher Scientific, catalog number: K0503 )
- Zymolyase® 100T from Arthrobacter leuteus (Amsbio LLC, catalog number: 120493-1 )
- One Shot® TOP10 chemically competent E. coli (Life Technologies, InvitrogenTM, catalog number: C4040-03 )
Note: These serve as the seed cultures for further chemically competent procedure using CCMB80 buffer. Our competent cells are prepared by inoculating a 1 L culture of SOB medium (Hanahan et al., 1991) with One Shot® TOP10 cells and growing them to A600 nm~0.3 at 23 °C. After harvesting, the TOP10 cells were made chemically competent by treating them, as described (Hanahan et al., 1991), with “CCMB80 buffer” (10 mM KOAc, pH 7.0, 80 mM CaCl2.2H2O, 20 mM MnCl2.4H2O, 10 mM MgCl2.6H2O, 10% glycerol), which was adjusted to pH 6.4 with 0.1 N HCl (if necessary), filter sterilized, and stored at 4 °C. After treatment, the competent cells were stored in aliquots at -80 °C. (Different ultra-chemically competent E. coli strains may be used in place of TOP10 for this procedure). - Ampicillin (final concentration of 100 μg/ml; Research Products International Corp., catalog number: A40040-100.0 ) and Kanamycin (final concentration of 50 μg/ml; Life Technologies, catalog number: 11815-024 )
- 1 M sorbitol, 0.1 M Na2EDTA (see Recipes)
- YPD liquid media (see Recipes)
- SOB medium (see Recipes)
- SOC medium (see Recipes)
- LB plates (with appropriate drug included) (see Recipes)
Equipment
- 0.5 mm glass beads (BioSpec Products, catalog number: 11079105 )
- Centrifuge (Eppendorf microcentrifuge model: 5415D , catalog number: 022621408 ; Eppendorf rotor, model: F-45-24-11 for 24 x 1.5/2 ml, catalog number: 022636502 )
- Petri dish (100 x 15 mm size; VWR International, catalog number: 25384-088 )
- Tube (Axygen Microtubes 1.5 ml clear, homo-polymer, boil-proof, catalog number: MCT-150-C )
- Vortexing adaptor (Microtube foam insert for Fisher Vortex Genie 2 mixer, Scientific Industries, Inc.; catalog number: 504-0234-00 )
- PCR machine (MJ Research PTC-200 Peltier Thermo Cycler, dual 30-well alpha blocks)
Procedure
- To begin, oligonucleotides are designed that allow for the amplification of each of the necessary gene fragment(s), epitope or fluorescent protein tags, nutritional markers, and/or drug-resistance cassettes and their recombinational assembly in the desired order (Figure 1B). The latter objective is achieved by "tails" of homology [(typically 30 nucleotides (Baudin et al., 1993)] on each primer that permit the joining of each amplified fragment to its neighbors by HR. Overlap sequences of 30 nucleotides in the 5'-tail on the N-terminal-most fragment and in the 3'-tail of the C-terminal-most fragment with the corresponding flanking sequences in the plasmid DNA also suffice for recombinational insertion into the vector.
- The desired fragment sequences can be amplified using as the template either genomic or plasmid DNA (see step 3) as a template. For purified plasmid DNA from most commercial kits (see Materials and Reagents), less than 0.25 μl is required as template DNA; for chromosomal DNA, approximately 2-3 μl is recommended per 50 μL PCR reaction using standard genomic DNA preparations from yeast (Amberg et al., 2006). We have found that optimal production of PCR fragments with overhanging tails is achieved using a two-phase PCR procedure: 5 cycles at a lower annealing temperature, followed by 25 cycles at a higher annealing temperature. To determine the initial temp., the Tm (in °C) for the non-overhanging portion of each of the two primers is determined (for this purpose, we use an algorithm available on-line (http://www.basic.northwestern.edu/biotools/oligocalc.html) and the recommended initial annealing temperature is determined from the formula [(Tm forward + Tm reverse) / 2] - 5. To determine the second temp., Tm's for each of the full primers are determined and the same formula applied. Depending on the DNA polymerase, the sequences being amplified, the thermocycler, and even the tubes used, the PCR reactions may need to be optimized. The critical point is that a single clean PCR product of the expected length needs to be generated. Hence, a sample of the resulting PCR product should be examined on a DNA agarose gel to ensure (i) a product of the correct size was created and (ii) there are not multiple DNA bands corresponding to mispriming.
- If any sequence amplification used as the template a yeast-based plasmid propagated in E. coli, subsequent digestion with DpnI (New England Biolabs) allows for destruction of the methylated template DNA and virtually eliminates the recovery of potential false positives in those cases where a marker being selected in the constructed vector of interest is the same as that contained in the template DNA. Digestion with DpnI is performed at 37 °C overnight (no heat-inactivation required) directly in the PCR tube mixture (no buffer required).
- The desired yeast vector of interest (Figure 1B) should be digested with a single unique restriction endonuclease; this site should be downstream of any inserted fragment that is already present in the vector. One strategy includes cloning beforehand a promoter sequence (5’-UTR) into the desired yeast-based plasmid (we prefer CEN vectors).
- Digestion of the resulting vector on the 3’-end of the promoter should be performed using an appropriate restriction enzyme and incubated overnight.
Note: This digested product does not have to be extracted or gel purified because even uncut vector will not yield any false positives whenever the marker for selection of the desired construct (e.g., hygromycin-resistance) is distinct from any marker in the parent plasmid (e.g., URA3) (however, nonetheless, it might be prudent to ensure that the plasmid has indeed been linearized by running a small sample on an agarose gel). - A yeast strain that results in efficient assembly and subsequent high yield of the desired plasmid DNA is SF838-1Dα (MATα ura3-52 leu2-3,122 his4-519 ade6 pep4-3 gal2) (Rothman and Stevens, 1986). However, for construction of plasmids in which HIS3 will be the selected marker (and not hygromycin-resistance), a derivative THS4218 (SF838-1Dα HIS4 his3Δ::HygR) is available. We have found that isolation of plasmids from strains of this background reproducibly results in higher plasmid yields and is more reliable than from other laboratory strains, including BY4741 (MATa leu2Δ ura3Δ met15Δ his3Δ) and its derivatives (Brachmann et al., 1998), especially when combined with ultra-chemically competent TOP10 bacterial transformation (see below).
- The yeast should be grown overnight to saturation in rich (YPD) medium at 30 °C, back-diluted to A600 nm ~0.25 in 10 ml of YPD and grown at 30 °C with shaking for roughly 4-5 h until A600 nm ~1. For each plasmid construction by this in vivo ligation procedure, a 10 ml culture of yeast is required. Hence, if multiple plasmid constructions are to be done in parallel, then commensurately more culture will be needed.
- Yeast should be collected by sedimentation in a clinical centrifuge for 5 min at 6,000 rpm and the YPD medium poured off. Resuspend the resulting pellet in 500 μl of sterile water with gentle pipetting and sediment again in a microfuge for 1 min at 6,000 rpm.
- For yeast transformation, a modified lithium acetate-based high-efficiency protocol should be used (Eckert-Boulet et al., 2012). For this purposed, remove the water with a pipet and resuspend the yeast pellet in 500 μl of sterile 100 mM lithium acetate solution and re-collect the cells for 1 min at 6,000 rpm and remove the supernatant solution and keep the yeast pellet.
- To each yeast pellet, add the following to the tube: 240 μl of 50% polyethylene glycol (PEG; autoclaved and sterile), 36 μl of sterile 1 M lithium acetate, 50 μl of 10 mg/ml single-stranded salmon sperm DNA (ssDNA in water). A high amount of ssDNA is included to (i) serve as an efficient carrier of the transformed DNA and (ii) the ssDNA is extremely viscous (and can clump into globs that resist transferring via micropipette) at concentrations of 10 mg/ml and therefore, a significant amount is lost in pipetting the solution into each tube (or a master mix solution). Therefore, to ensure that adequate ssDNA is included, we suggest the approximate amount of 50 μl with the understanding that some amount will be lost in solution preparation. Ingredients should be added in this order with the PEG solution added to the yeast first. Alternatively, should multiple transformations be performed at the same time, a “master mix” of the PEG, lithium acetate, and ssDNA can be mixed together and vortexed vigorously for 30 sec prior to adding to each yeast pellet. Two aspects of these reagents are important. First, the stock of salmon sperm DNA should be boiled for 10 min and cooled on ice for 10 min every time that it is used. Second, if the "master mix" is used, it must be made fresh for each experiment (it cannot be stored and reused).
- Add the appropriate DNA fragments (15-20 μl from each PCR reaction; the concentration will be dependent on the efficiency of the PCR reaction the length of the fragment, but generally this corresponds to roughly 3-5 μg of each PCR reaction) and cleaved vector (15-20 μl from the restriction enzyme digest; and approximately 2 μg total DNA content depending on the size of the parent vector being used) to the tube of lithium acetate- and PEG-treated yeast. Vortex the tube for 30 sec at maximum speed to resuspend the yeast into solution and incubate at exactly 42 °C for 45 min.
- Collect the yeast for 1 min at 6,000 rpm in a microfuge, remove the supernatant solution, and gently resuspend the yeast pellet in 550 μl sterile YPD medium and incubate overnight standing in a rack in a 30 °C incubator. This overnight incubation step is absolutely required for any constructs using a drug-resistance cassette (KanR, NatR or HygR); yeast require a sufficient amount of time to express the drug-resistance cassette and become resistant before they are challenged with the drug present in the selection medium. Standing overnight incubation, as opposed to aeration by shaking or rotation, allows for slow and largely anaerobic growth reducing the rate of production of CO2 and other volatiles, which, in our experience, can pop the lids off of standard Eppendorf tubes.
- Plate the full tube of yeast on the appropriate agar medium (YPD + drug, or synthetic drop-out medium), and incubate the plates for at least two days at 30 °C. Optional controls include transforming the same yeast strains with (i) only digested vector and/or (ii) only each PCR fragment-both of these should result in no plasmid constructs being generated and no growth on the selection plates.
- For recovery of plasmid DNA, scrape yeast off the plate (avoid agar)-use at least 25% of the surface area of the entire Petri dish (150 mm diameter dish; assuming there is a full lawn of coverage). Ideally, this high-efficiency in vivo ligation technique coupled with the high-efficiency yeast transformation should result in a complete lawn on the selective medium, although other factors (such as the complexity of the assembly) can reduce the final colony count. It is advised to only proceed with the protocol with constructs that resulted in ample coverage on the selection plate. If there are only single colonies on the initial selection plate, it is recommended to collect all of them and replate onto a fresh (selective) plate to grow up a sufficiently dense lawn of yeast for use in this protocol.
- The yeast scraped from the plate are resuspended in 500 μl of 1 M sorbitol, 0.1 M Na2EDTA (pH 7.5) (Amberg et al., 2006), briefly vortexed (maximum speed) to ensure full resuspension (expect that the solution will be very thick and concentrated), and then 5-7.5 μl of a stock (final concentration of 25 mg/ml in 50% glycerol) of Zymolyase 100T (stored at 4 °C) is added. After incubation at 37 °C on rotator for at least 1 h (but not more than 2 h), the resulting spheroplasts are collected at 12,000 rpm in a microfuge.
- After removal of the supernatant solution, the spheroplast pellet is resuspended in 250 μl of the proprietary bacterial plasmid isolation solution (see Materials and Reagents). Triturate by drawing the solution in and out of a pipet; at this point, the solution of lysed spheroplasts is extremely viscous and it takes significant effort to ensure very thorough resuspension.
- Add a roughly equal volume (roughly 100-250 μl) of glass beads (0.5 mm diameter). Briefly spin in microfuge at 2,000 rpm for 5 sec (to ensure all glass beads are mixed with the yeast solution rather than stuck to the walls of the tube). Vortex on maximum for 5 min (if you are doing multiple constructions in parallel, a vortexing adaptor that can house multiple tubes at once can be used).
- After completion of lysis by the glass bead breakage, proceed with the subsequent steps of your preferred commercial bacterial plasmid isolation protocol- for the Thermo Scientific system, add 250 μl of “Lysis buffer” and mix by inversion at least 10 times= and incubate at room temperature for 5 min. Add 350 μl of “Neutralization buffer” and mix by inversion at least 10 times immediately. Subject the resulting solution to centrifugation at maximum (13,200 rpm) in microfuge for 5 min.
- Transfer by pipetting the supernatant DNA-containing solution into a fresh, labeled tube and centrifuge at maximum speed for an additional 2 min (to remove any residual precipitate/cell debris). Transfer by pipetting the clarified supernatant solution into the plasmid-capturing spin column provided in the commercial bacterial plasmid isolation kit (see Materials and Reagents), centrifuge at maximum speed for 1 min and discard the flow-through.
- Wash the spin column with the commercial ethanol wash solution (500-750 μl), centrifuge at maximum speed for 1 min, and discard flow-through. Repeat (although this second wash step varies with the commercial plasmid prep kits and may be optional).
- To remove any residual wash solution, place the empty spin column in the microfuge and spin at maximum speed for 1 min, then place the spin column in a fresh Eppendorf tube and elute the bound DNA in the spin column by adding 25-30 μl of the “elution buffer” provided by the commercial supplier, incubating 1-2 min at room temperature, and then collecting the eluate by centrifugation at maximum speed for 2 min.
- To recover the desired constructed plasmid, a sample (10 μl) of the DNA-containing solution eluted from the spin column is gently mixed in a tube containing 100 μl of ultrachemically competent TOP10 bacterial cells (Materials and Reagents) that was taken from the -80 °C freezer and thawed on ice for ten minutes. After combining the DNA with the competent cells, the solution was left on ice for another ten min, then placed in a 42 °C water bath for 40 sec to subject the cells to a heat shock, returned to ice for two additional min, mixed with 500 μl of sterile SOC medium, and incubated on a rotator at 37 °C for 1 h.
- The resulting bacterial transformants (50-100 μl of the bacterial culture in SOC medium) are plated onto appropriate LB + drug (typically ampicillin or kanamycin) plates (prewarmed at 37 °C) and incubated at 37 °C overnight. Typically, 100-500 single colonies per plate grow out. Plasmid DNA from several representative single colonies are then isolated by standard molecular biology methods and tested using diagnostic PCR and DNA sequencing to ensure that the assembled gene fragments are all present and do not harbor any mutations or other alterations. This overall in vivo ligation process is so incredibly robust and efficient, it usually only requires 1-2 colonies to be analyzed to obtain the desired construct (see Notes).
- Important advice for avoiding pitfalls: Application of the above procedures for a multitude of different constructs (n > 1,000) over many years (a decade) indicates that the most common cause of any difficulty in successfully obtaining the desired constructs arises from the degree of sequence impurity in commercially purchased synthetic oligonucleotides. We have found that when mutations are observed, they reside in the junctions between gene fragments that correspond directly to the primer sequences used in the initial PCR amplifications. Thus, ensuring the high purity/quality/homogeneity of the initial oligonucleotide primers is critically important for maximizing the efficiency of this method of DNA assembly.
Notes
Our system includes a number of important differences from a previous description (Andersen, 2011) of this general approach for HR-driven fragment assembly and plasmid construction in yeast.
- Our methodology for plasmid creation takes advantage of the incorporation of a PCR fragment that contains a selectable marker (auxotrophic or drug-resistance cassette) that is distinct from any which may reside on any of the template DNAs used or on the parent vector into which the construct will be inserted (Figure 1B). In this way, even if the vector recircularizes, it will not generate any false positives when the selection is imposed to isolate clones carrying the desired construct.
- Whenever it is unavoidable that the template and the vector have the same selectable marker, in our procedure false positives can still be efficiently eliminated by treatment of the PCR reaction products with DpnI restriction enzyme before their incorporation into the vector.
- Our method eliminates the use of phenol: chloroform in the yeast plasmid isolation step.
- Use of a unique lineage of S. cerevisiae strains (SF838-1Dα-based) allows for more efficient plasmid rescue from yeast.
- Our approach allows for the insertion at any location of any desired sequence wherever it may have utility for meeting any experimental goal. Such sequences could include, but are not limited to, at the protein level, internal epitope tags, protease cleavage sites, aptamers for protein recognition domains, flexible linkers, and subcellular localization signals and, at the nucleic level, restriction endonuclease cut sites, stem-loop sequences for RNA-binding proteins, loxP cassettes, etc. (Figure 1C).
- Our approach exploits and highlights the power of in vivo HR to yield complex multi-part gene fusions, domain deletions, or tagged constructs in a single assembly step. Prior to our methods, this approach had only been used as a means to simply clone by reassembly a large contiguous stretch of the yeast genome (Andersen, 2011).
- Routinely, even for plasmids constructed from 5 or more separate PCR fragments (with a final plasmid size of 7-15 kb), only 1-2 bacterial clones need to be examined to obtain the construct of interest. Overall, the process of in vivo ligation and plasmid recovery described here is an efficient, cost-effective means for accurately generating complex DNA assemblies and can be applied in a massively parallel manner, when needed, to generate hundreds of plasmid constructs simultaneously.
- Finally, because a selectable marker is incorporated into each construct, it can be amplified (or excised) from the vector containing it and used for one-step gene replacement (Rothstein, 1991) in any desired recipient organism, such as the yeast KanR-marked haploid or diploid genome deletion collections (Brachmann et al., 1998) (Figure 1D).
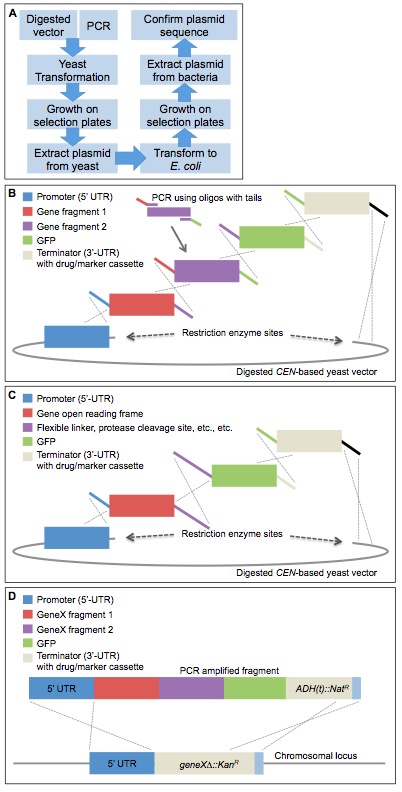
Figure 1. Complex DNA assembly using in vivo ligation by homologous recombination in yeast. A. A diagram illustrating the workflow of the protocol to construct plasmids using homologous recombination in yeast. B. An example construct is illustrated using homologous overlapping primer tails (identical colors show homology and the dotted lines illustrate the resulting recombination event across the homologous segments). Digested CEN-based yeast vector, such as LEU2-based pRS315 (Sikorski and Hieter, 1989). This illustration represents a single example of a series of gene fragments assembled into a single construct in one step using in vivo ligation. Modifications to this arrangement can be used to assemble multiple domains within a single gene or genes, include multiple epitope or fluorescent tags at either the N- or C-termini, and/or include flanking UTR sequences for subsequent integration at a non-native chromosomal locus. C. This example specifically addresses a useful property of this process. Specifically, one can encode in the homologous overlapping tails between two adjacent gene fragments to be assembled any desired non-native sequence information to introduce additional sequences of interest (such as, but not limited to, an epitope tag, a Gly-Ser rich linker sequence, restriction enzyme cut sites, protein cleavage motifs, signal sequences, etc.). D. Following construction, the entire construct can be PCR-amplified and integrated into the genome in a single step using either designed homology (5’- and 3’-UTR) within the system or, in the case of the MX-4-based drug-resistance cassette system devised by Goldstein and McCusker (2001), homology between the common sequence (blue) installed downstream of each type of drug-resistance gene, in which ADH1(t) is the terminator sequence from the ADH1 locus. Replacement of the gene deletion cassette with the modified gene of interest also allows for a swap in the growth selection markers (either drug resistance or nutrient). This system can also be modified to accommodate essential genes, as long as the recipient cell also harbors a URA3-based vector expressing the WT essential gene from a second plasmid, which can be subsequently eliminated by counter-selection on 5-FOA medium (Boeke et al., 1984).
Recipes
- 1 M sorbitol, 0.1 M Na2EDTA
Adjusted to pH 7.5, filter sterilized - YPD liquid media
1% yeast extract
2% peptone
2% dextrose
Autoclaved - SOB medium
2% tryptone
0.5% yeast extract
10 mM NaCl
2.5 mM KCl
1 mM MgCl2
1 mM MgSO4 - SOC medium
SOB medium
20 mM glucose final concentration - LB plates (with appropriate drug included)
15 g/L agar
1% tryptone
0.5% yeast extract
10 g/L NaCl
4 mM NaOH
Acknowledgments
This work was supported by a Miller Research Fellowship from the Miller Institute for Basic Research in Science at the Univ. of California, Berkeley (to G.C.F.) and by NIH Research Grants GM21841 (to J.T.) and GM101314 (to Eva Nogales and J.T.). This protocol was adapted and modified from Finnigan et al. (2011).
References
- Amberg, D. C., Burke, D. J. and Strathern, J. N. (2006). Yeast DNA isolation: miniprep. CSH Protoc 2006(1).
- Andersen, E. C. (2011). PCR-directed in vivo plasmid construction using homologous recombination in baker's yeast. Methods Mol Biol 772: 409-421.
- Anderson, P. R. and Haj-Ahmad, Y. (2003). Counter-selection facilitated plasmid construction by homologous recombination in Saccharomyces cerevisiae. Biotechniques 35(4): 692-694, 696, 698.
- Baudin, A., Ozier-Kalogeropoulos, O., Denouel, A., Lacroute, F. and Cullin, C. (1993). A simple and efficient method for direct gene deletion in Saccharomyces cerevisiae. Nucleic Acids Res 21(14): 3329-3330.
- Boeke, J. D., LaCroute, F. and Fink, G. R. (1984). A positive selection for mutants lacking orotidine-5'-phosphate decarboxylase activity in yeast: 5-fluoro-orotic acid resistance. Mol Gen Genet 197(2): 345-346.
- Brachmann, C. B., Davies, A., Cost, G. J., Caputo, E., Li, J., Hieter, P. and Boeke, J. D. (1998). Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast 14(2): 115-132.
- Eckert-Boulet, N., Pedersen, M. L., Krogh, B. O. and Lisby, M. (2012). Optimization of ordered plasmid assembly by gap repair in Saccharomyces cerevisiae. Yeast 29(8): 323-334.
- Finnigan, G. C., Hanson-Smith, V., Houser, B. D., Park, H. J. and Stevens, T. H. (2011). The reconstructed ancestral subunit a functions as both V-ATPase isoforms Vph1p and Stv1p in Saccharomyces cerevisiae. Mol Biol Cell 22(17): 3176-3191.
- Giaever, G., Chu, A. M., Ni, L., Connelly, C., Riles, L., Veronneau, S., Dow, S., Lucau-Danila, A., Anderson, K., Andre, B., Arkin, A. P., Astromoff, A., El-Bakkoury, M., Bangham, R., Benito, R., Brachat, S., Campanaro, S., Curtiss, M., Davis, K., Deutschbauer, A., Entian, K. D., Flaherty, P., Foury, F., Garfinkel, D. J., Gerstein, M., Gotte, D., Guldener, U., Hegemann, J. H., Hempel, S., Herman, Z., Jaramillo, D. F., Kelly, D. E., Kelly, S. L., Kotter, P., LaBonte, D., Lamb, D. C., Lan, N., Liang, H., Liao, H., Liu, L., Luo, C., Lussier, M., Mao, R., Menard, P., Ooi, S. L., Revuelta, J. L., Roberts, C. J., Rose, M., Ross-Macdonald, P., Scherens, B., Schimmack, G., Shafer, B., Shoemaker, D. D., Sookhai-Mahadeo, S., Storms, R. K., Strathern, J. N., Valle, G., Voet, M., Volckaert, G., Wang, C. Y., Ward, T. R., Wilhelmy, J., Winzeler, E. A., Yang, Y., Yen, G., Youngman, E., Yu, K., Bussey, H., Boeke, J. D., Snyder, M., Philippsen, P., Davis, R. W. and Johnston, M. (2002). Functional profiling of the Saccharomyces cerevisiae genome. Nature 418(6896): 387-391.
- Gibson, D. G. (2011). Enzymatic assembly of overlapping DNA fragments. Methods Enzymol 498: 349-361.
- Goldstein, A. L. and McCusker, J. H. (1999). Three new dominant drug resistance cassettes for gene disruption in Saccharomyces cerevisiae. Yeast 15(14): 1541-1553.
- Gunyuzlu, P. L., Hollis, G. F. and Toyn, J. H. (2001). Plasmid construction by linker-assisted homologous recombination in yeast. Biotechniques 31(6): 1246, 1248, 1250.
- Hanahan, D., Jessee, J. and Bloom, F. R. (1991). Plasmid transformation of Escherichia coli and other bacteria. Methods Enzymol 204: 63-113.
- Iizasa, E. and Nagano, Y. (2006). Highly efficient yeast-based in vivo DNA cloning of multiple DNA fragments and the simultaneous construction of yeast/ Escherichia coli shuttle vectors. Biotechniques 40(1): 79-83.
- Joska, T. M., Mashruwala, A., Boyd, J. M. and Belden, W. J. (2014). A universal cloning method based on yeast homologous recombination that is simple, efficient, and versatile. J Microbiol Methods 100: 46-51.
- Ma, H., Kunes, S., Schatz, P. J. and Botstein, D. (1987). Plasmid construction by homologous recombination in yeast. Gene 58(2-3): 201-216.
- Muhlrad, D., Hunter, R. and Parker, R. (1992). A rapid method for localized mutagenesis of yeast genes. Yeast 8(2): 79-82.
- Oldenburg, K. R., Vo, K. T., Michaelis, S. and Paddon, C. (1997). Recombination-mediated PCR-directed plasmid construction in vivo in yeast. Nucleic Acids Res 25(2): 451-452.
- Rothman, J. H. and Stevens, T. H. (1986). Protein sorting in yeast: mutants defective in vacuole biogenesis mislocalize vacuolar proteins into the late secretory pathway. Cell 47(6): 1041-1051.
- Rothstein, R. (1991). Targeting, disruption, replacement, and allele rescue: integrative DNA transformation in yeast. Methods Enzymol 194: 281-301.
- Sikorski, R. S. and Hieter, P. (1989). A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 122(1): 19-27.
- Szostak, J. W., Orr-Weaver, T. L., Rothstein, R. J. and Stahl, F. W. (1983). The double-strand-break repair model for recombination. Cell 33(1): 25-35.
- Winzeler, E. A., Shoemaker, D. D., Astromoff, A., Liang, H., Anderson, K., Andre, B., Bangham, R., Benito, R., Boeke, J. D., Bussey, H., Chu, A. M., Connelly, C., Davis, K., Dietrich, F., Dow, S. W., El Bakkoury, M., Foury, F., Friend, S. H., Gentalen, E., Giaever, G., Hegemann, J. H., Jones, T., Laub, M., Liao, H., Liebundguth, N., Lockhart, D. J., Lucau-Danila, A., Lussier, M., M'Rabet, N., Menard, P., Mittmann, M., Pai, C., Rebischung, C., Revuelta, J. L., Riles, L., Roberts, C. J., Ross-MacDonald, P., Scherens, B., Snyder, M., Sookhai-Mahadeo, S., Storms, R. K., Veronneau, S., Voet, M., Volckaert, G., Ward, T. R., Wysocki, R., Yen, G. S., Yu, K., Zimmermann, K., Philippsen, P., Johnston, M. and Davis, R. W. (1999). Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science 285(5429): 901-906.
Article Information
Copyright
© 2015 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Finnigan, G. C. and Thorner, J. (2015). Complex in vivo Ligation Using Homologous Recombination and High-efficiency Plasmid Rescue from Saccharomyces cerevisiae. Bio-protocol 5(13): e1521. DOI: 10.21769/BioProtoc.1521.
-
Finnigan, G. C., Hanson-Smith, V., Houser, B. D., Park, H. J. and Stevens, T. H. (2011). The reconstructed ancestral subunit a functions as both V-ATPase isoforms Vph1p and Stv1p in Saccharomyces cerevisiae. Mol Biol Cell 22(17): 3176-3191.
Category
Microbiology > Microbial genetics > DNA
Molecular Biology > DNA > DNA cloning
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.