- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

DNA Damage Sensitivity Assays in Caenorhabditis elegans

Published: Vol 5, Iss 11, Jun 5, 2015 DOI: 10.21769/BioProtoc.1487 Views: 13107
Reviewed by: Peichuan ZhangMichael EnosMohlopheni Marakalala
Original research article
The authors used this protocol in:

Oct 2014
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
C. elegans has served as a genetically tractable multicellular model system to examine DNA damage-induced genotoxic stress which threatens genome integrity. Importantly, the high degree of conservation shared between worms and humans offers the advantage that findings about DNA damage-induced cell cycle arrest/checkpoint response and DNA double-strand break repair in worms are applicable to human studies. Here, we describe simple DNA damage sensitivity assays to quantify the response of C. elegans to diverse types of DNA damaging agents. These assays have provided important insights into the mechanisms of function for factors such as ZTF-8 that are involved in DNA damage repair and response in the C. elegans germline. These DNA damage sensitivity assays rely on the straightforward readouts of either egg or larval lethality and involve the use of various DNA damaging agents. We use γ-irradiation (γ-IR), which produces DNA double-strand breaks (DSBs), camptothecin (CPT), which induces single-strand breaks, nitrogen mustard (HN2), which produces interstrand crosslinks (ICLs), hydroxyurea (HU), which results in replication fork arrest thus preventing DNA synthesis, and UV-C, which causes photoproducts (pyrimidine dimers). See Table 1. Comparisons between the relative sensitivity/resistance observed in, for example, mutants compared to wild type, for various DNA damaging agents allows for inferences regarding potential repair pathways being affected.
Keywords: DNA damageMaterials and Reagents
- E.coli OP50 (Carolina, catalog number 155073 )
- M9 (He, 2011)
- NGM agar media (He, 2011)
- M9 500 ml + Triton X-100 50 μl
- Mechlorethamine hydrochloride (Sigma-Aldrich, catalog number 122564 )
- Camptothecin (Sigma-Aldrich, catalog number C9911 )
- Hydroxyurea (Sigma-Aldrich, catalog number H8627 )
- Triton X-100 (Sigma-Aldrich, catalog number T8787 )
- 20% Alkaline Hypochlorite Solution (see Recipes)
Equipment
- 24-well plates (OLYMPUS, catalog number 25-102 )
- Short tip disposable glass Pasteur pipets (VWR International, catalog number 14673-010 )
- Petri dishes (60 x 15 mm) (VWR International, catalog number 25384-092 )
- Sealing film (Parafilm M, catalog number: PM996 )
- Nutator mixer (Clay Adams, model: 1105 Mixer )
- 20 °C incubator (Thermo Fisher Scientific, Precision 815)
- 37 °C shaking incubator (New Brunswick, Innova 4330 )
- 37 °C incubator ((VWR Internationa, 1510E)
- Benchtop centrifuge for spinning 15 ml tubes (Lanmet, Hermle Z, model: 400K )
- Stereo microscope (Leica Microsystems, model: MZ75 )
- UV crosslinker (Stratagene, model: 2400 Stratalinker) with 254 nm UV bulbs
- γ-IR irradiator (Shepherd & Associates, Mark 1 Cs137 irradiator)
Procedure
- Preparation of genotoxins
DNA damaging agents are generally toxic and require extra safety precautions when working with them. Wear double gloves and a lab coat. Always work in a laminar flow and label and seal your containers in hard-walled robust containers. Make fresh solutions to avoid possible degradation and always seal the tubes, even for short-term storage. Caution must be exercised to assure that all waste materials are disposed of properly.
In this protocol, solid/agar media is used for UVC, γ-IR, and HU sensitivity assays, while CPT and HN2 treatments are performed in liquid culture (Table 1). Defects in meiotic pachytene are addressed by analysis 26-28 h post exposure (Jaramillo-Lambert et al., 2010). Mitotic repair failures are examined by analysis 48-72 h post exposure.- Pyrimidine dimmers: UV irradiation treatment can be performed utilizing the 2400 Stratalinker. Worms are exposed to 0 to 150 J/m2 of UVC. The lids are removed from the Petri dishes to ensure better UV penetration. Most UV crosslinkers have a UV sensor inside so caution must be taken to avoid blocking/disrupting the UV sensor by placing plates next to it. After treatment, worm plates are wrapped in foil to avoid potential photo recovery. Known sensitive strains: slx-1 (tm2644) (Saito et al., 2012) and him-18 (tm2181) (Saito et al., 2012).
- DNA double-strand breaks: To assess for γ-IR sensitivity, doses ranging between 0 to 150 Gy are generally used. Find the radiation dose rate (Gy/h) for your irradiator to calculate a time for exposure. Known sensitive strains: him-18 (tm2181) (Saito et al., 2012), brc-1 (tm1145) (Ward et al., 2007), and ztf-8 (tm2176) (Kim and Colaiacovo, 2014).
- Replication arrest: Hydroxyurea is soluble in water. Dissolve HU completely with a good amount of water (~10 ml) and mix it into autoclaved NGM agar media (1 L) when it has cooled down to 55-50 °C. HU is hygroscopic and must be sealed and stored in a desiccator. HU sensitivity is assessed by placing animals on NGM plates seeded with OP50 (E.coli) and containing 0 to 40 mM HU for 20 h. Known sensitive strains: clk-2 (mn159) (Bailly et al., 2010),and ztf-8 (tm2176) (Kim and Colaiacovo, 2014).
- Single-strand breaks: Camptothecin is not soluble in water and instead is soluble in DMSO at 10 mg/ml. Higher concentrations require heat for it to completely enter into solution (10 min at 95 °C). It is advisable to prepare a lower concentration to avoid heating a toxic material. The final range of concentrations used for camptothecin is 0 to 1,000 nM in M9 buffer containing OP50. M9 solution at a pH of 6.0 was reported to deliver higher sensitivity compared with pH 7.0 by way of better impeding topoisomerase activity (Kessler and Yanowitz, 2014). Known sensitive strains: him-18 (tm2181) (Saito et al., 2012) and brc-1 (tm1145) (Ward et al., 2007).
- Interstrand cross-links: Nitrogen mustard (HN2) is soluble in water. For nitrogen mustard (HN2) sensitivity, animals are treated with 0 to 150 μM of HN2 in M9 buffer containing OP50. Known sensitive strains: brc-1 (tm1145) (Ward et al., 2007) and slx-1 (tm2644) (Saito et al., 2012).
Table 1. Known DNA damage sensitive strains for genotoxins. A summary of genotoxins, the lesions they produce, and sensitive strains. The media used for this protocol is also indicated.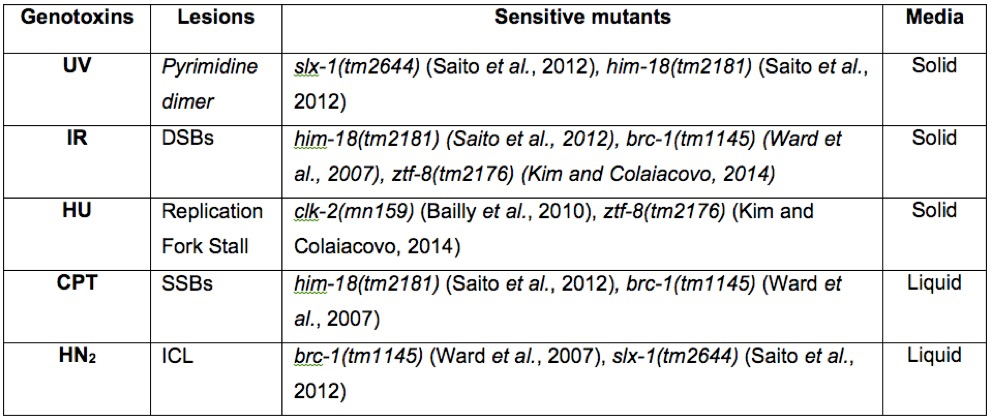
- Pyrimidine dimmers: UV irradiation treatment can be performed utilizing the 2400 Stratalinker. Worms are exposed to 0 to 150 J/m2 of UVC. The lids are removed from the Petri dishes to ensure better UV penetration. Most UV crosslinkers have a UV sensor inside so caution must be taken to avoid blocking/disrupting the UV sensor by placing plates next to it. After treatment, worm plates are wrapped in foil to avoid potential photo recovery. Known sensitive strains: slx-1 (tm2644) (Saito et al., 2012) and him-18 (tm2181) (Saito et al., 2012).
- Worm preparation
C. elegans strains are cultured in NGM plates (60 x 15 mm petri dishes) seeded with OP50 E. coli at 20 °C under standard conditions as described in Brenner (1974). In brief, 10 ml NGM agar containing plates are prepared and dried in storage bins kept at room temperature for 4-5 days before being seeded with OP50 E. coli and placed overnight in a 37 °C incubator. The N2 Bristol strain is used as the wild-type control together with sensitive mutants as described in Table 1. - Daily procedures
Basic procedures for both liquid and solid exposures are similar, as illustrated in Figure 1, although the liquid exposures require additional washes before and after mutagen treatment. Calculate the total number of worms required for an assay. For example, for three doses of exposure, approximately 75 worms of each genotype are required (5 worms/plate x 5 plates x 3 doses). However, since some worms can be lost as they are prone to attaching to plastics, such as pipette tips and Eppendorf tubes, and given the potential lethality that may occur due to exposure to the genotoxins, it is recommended to have 2 times more worms in general.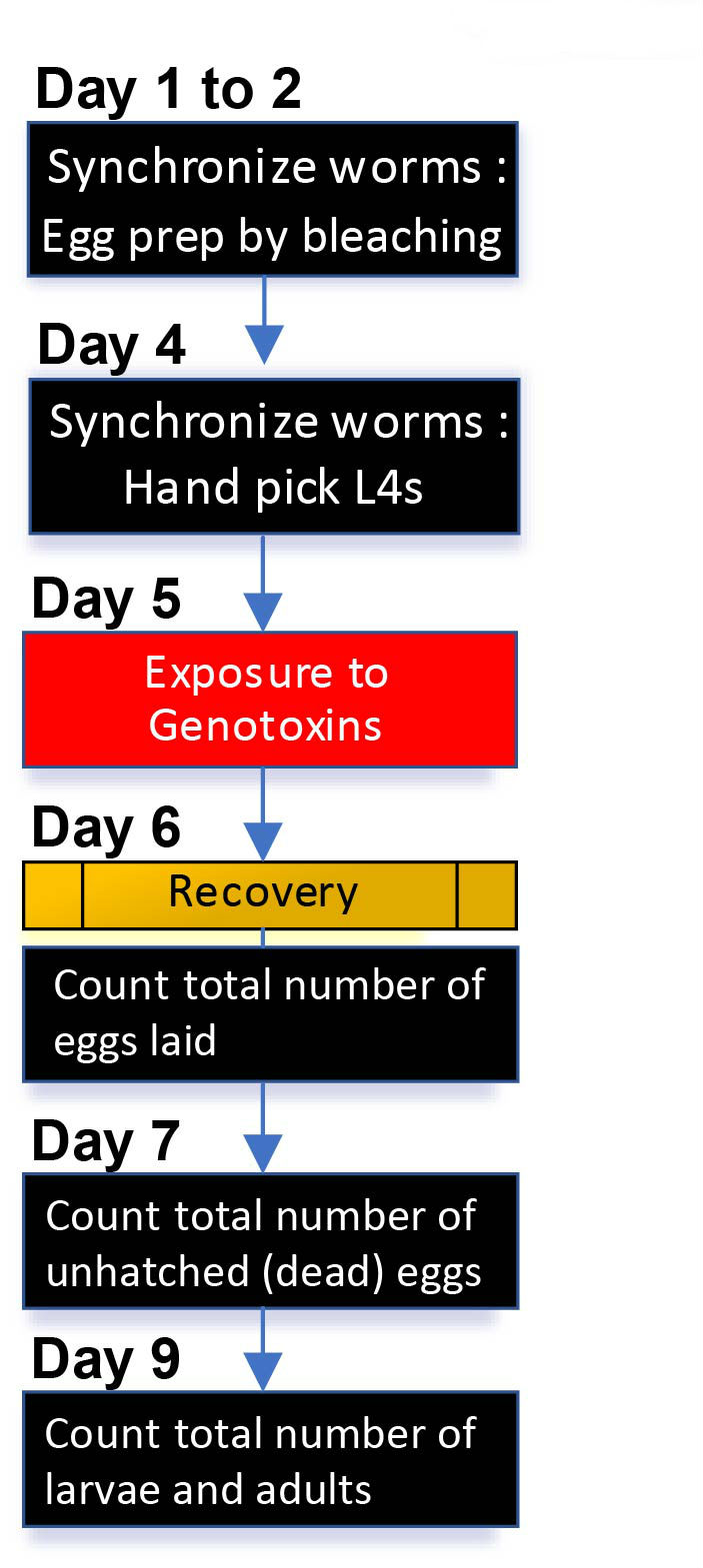
Figure 1. A schematic view of daily procedures for DNA damage sensitivity assay. Synchronized young adults are prepared by either egg prep by bleaching worms at day 1 or hand picking L4 worms at day 4. As illustrated, basic procedures for both liquid and solid exposures are similar. Note that liquid exposure requires additional washes before and after exposure to mutagens. See section B. Daily procedures for details.
Day 1- To prepare synchronized L1 stage larvae, gravid adult hermaphrodites are washed off from the plates by adding 1-2 ml of M9/plate. Leave the plates with M9 for 30-60 sec to dislodge the worms. Alternatively, for a small-scale test, L4 worms can be picked with a platinum wire instead of washing off the plate. However, synchronizing via bleaching removes potential fungal contamination of the worm which can be problematic when counting worms. The total number of adult worms required might vary, but 40 adults are required to obtain ~160 L1 larvae. If you have synchronized L4 worms ready for the assays, skip this step and start from step C10.
- Use a glass Pasteur pipette to transfer worms to 15 ml falcon tubes. (*Short tip length Pasteur pipettes are easier to manipulate. It is convenient to mark the 300 μl line on the 15 ml tubes used in step C3.)
- Spin the worms in a swing-bucket centrifuge (30-60 sec at ~1,000 rpm or ~210 rcf) and remove the supernatant by gently aspirating the M9. Leave ~ 300 μl of solution at the bottom to avoid disturbing worm pellets.
- Add 3 ml of 20% alkaline hypochlorite solution and gently swirl the tube. Most worms will break open and release their eggs in ~ 5 min. Do not bleach too long to avoid killing the eggs.
- Add ~15 ml of M9 to each tube and invert the tubes a few times.
- Repeat step C3 three more times to remove the alkaline hypochlorite solution.
- Add M9 up to 15 ml to the tubes and incubate overnight at 20 °C with gentle rocking (Nutator mixer). Seal the cap with Parafilm to avoid leaking.
- Repeat step C3.
- From the ~300 μl of solution remaining at the bottom following centrifugation and removal of the supernatant, distribute proper amount of liquid containing L1 worms to an OP50 seeded plate. Count the number of worms and make sure not to have either too many or not enough worms in a plate. Aim for ~50 worms for each seeded plate. If you start from 40 gravid adults, distribute 50 μl to 6 seeded plates (total 300 μl) after gently mixing the pellet by vortexing. Incubate ~48 h at 20 °C until worm reaching the L4 larval stage.
Briefly monitor worm growth. Make sure worms are not starved.
Day 4- Worms will be at the L4 stage. Incubate an additional 18-20 h and young adult worms will be ready for further analysis. In general, pick twice the number of worms for each dose per genotype. For example, pick 150 worms of N2 worms for three doses (25 worms x 3 doses x 2 times).
- For exposure in liquid media (CPT and HN2): Inoculate OP50 in 20 ml LB media in a 50 ml conical tube and incubate in a shaker incubator overnight at 37 °C. Note that a 20 ml culture is enough for 3 genotypes (two controls + sample) with 3 doses of exposures in liquid.
- For exposure in solid media (UV, γ-IR and HU): Age-matched young adults are transferred to OP50 seeded plates for γ-IR and UV exposures. Age-matched young adults are transferred to HU-containing seeded plate for HU sensitivity assay (Figure 2A).
For γ-IR and UV treatments, worms are exposed to the DNA damaging source and then incubated for 18-20 h at 20 °C. Go to step C19.
For HU treatment, worms are incubated in HU containing media for 18-20 h at 20 °C. Go to step C19. Avoid a thick bacterial lawn as it might affect UV/IR penetration. Only use plates seeded with overnight grown bacteria. See Notes.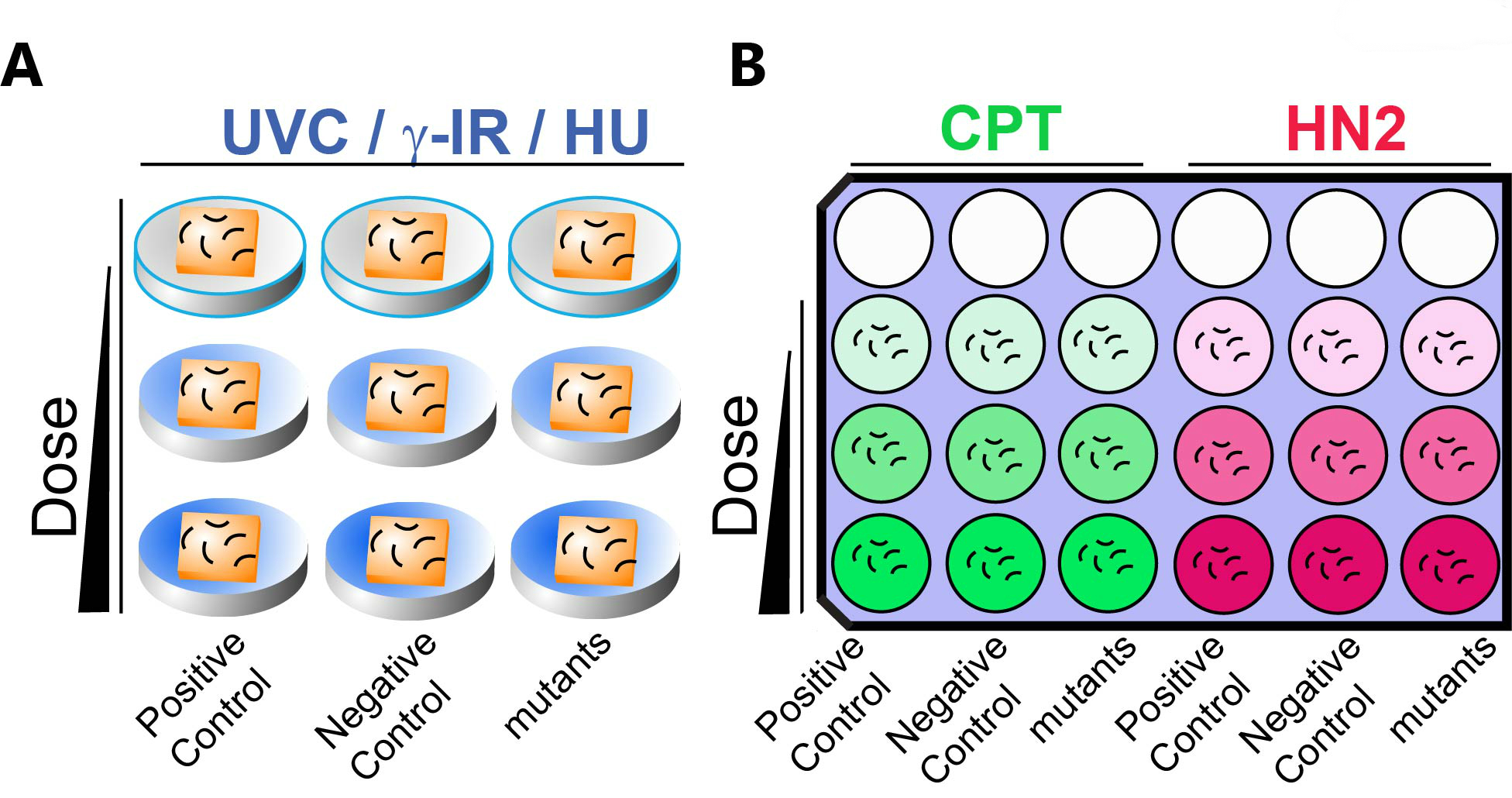
Figure 2. A schematic representation of DNA damage sensitivity assay design with solid (A) and liquid (B) exposure. A. For UVC, γ-IR and HU treatments, animals are exposed on seeded solid NGM media. B. One set of 9 wells is used for CPT (Green) or HN2 (Red) sensitivity. Animals are exposed to genotoxins suspended in liquid culture. Dose of genotoxin is indicated by the gradient color. Empty circles indicate empty wells. Positive (known sensitive mutants, see Table1) and negative controls (wild type N2) as well as mutant worms are incubated in the indicated column. Short curved lines represent animals in panels A and B. Squares represent bacterial lawns in panel A. - For exposure in liquid media (CPT and HN2): Spin down 20 ml of OP50 (OD600 >2.0) in LB media and remove supernatant. Add 4 ml of M9 and vortex to resuspend the pellet. For example, split 1.3 ml bacterial solution to 3 Eppendorf tubes for 3 doses of exposure including one for a mock exposure.
- Add genotoxin to the tubes containing OP50 for a 1.25x higher than the desired final concentration and Vortex briefly. For example, for final 100 μM, make 125 μM 1.3 ml HN2 + OP50.
- Dispensing genotoxins: Transfer 400 μl to each of 3 wells of a 24-well plate (horizontal direction). Repeat this step for each dose. See Figure 2B for an example of 3 doses x 3 genotypes x 2 genotoxins.
- Dispensing worms: Transfer age-matched young adults to 15 ml tubes and wash with 15 ml M9 + Triton X-100 two times followed by two washes with only M9.
Aspirate M9 gently and leave ~ 300 μl solution at the bottom to avoid disturbing worm pellets. Transfer 100 μl worm to each of 3 vertical wells from step C15 (100 μl x 3 wells, see Figure 2B). This will result in a total of 500 μl/well with the desired final concentration of genotoxin plus animals (400 + 100 μl). Repeat this step for each genotype. - Wrap the plate with foil and incubate at 20 °C for 18-20 h with gentle shaking.
- Repeat steps C2-3 twice.
- Transfer worms to seeded NGM plates. (Gently swirl the plates to spread any excess liquid so it can be more efficiently absorbed by the media. Alternatively, aspirate the liquid carefully without sucking up worms.) Allow for recovery by incubating for 3 h at 20 °C.
- Pick worms and split 5 per plate and incubate at 20 °C for ~4 h (5 plates/dose/genotype). Avoid a thick bacterial lawn as it makes the counting harder. See Notes.
- P0 worms can be removed and discarded or can be used for further analysis such as immunostaining or DAPI staining. Count the number of eggs laid (for wild type N2, 70-100 eggs are expected per plate). Draw a grid with a dark colored marker on a Petri dish lid, which you can place under your plate to facilitate scoring. See Figure 3.
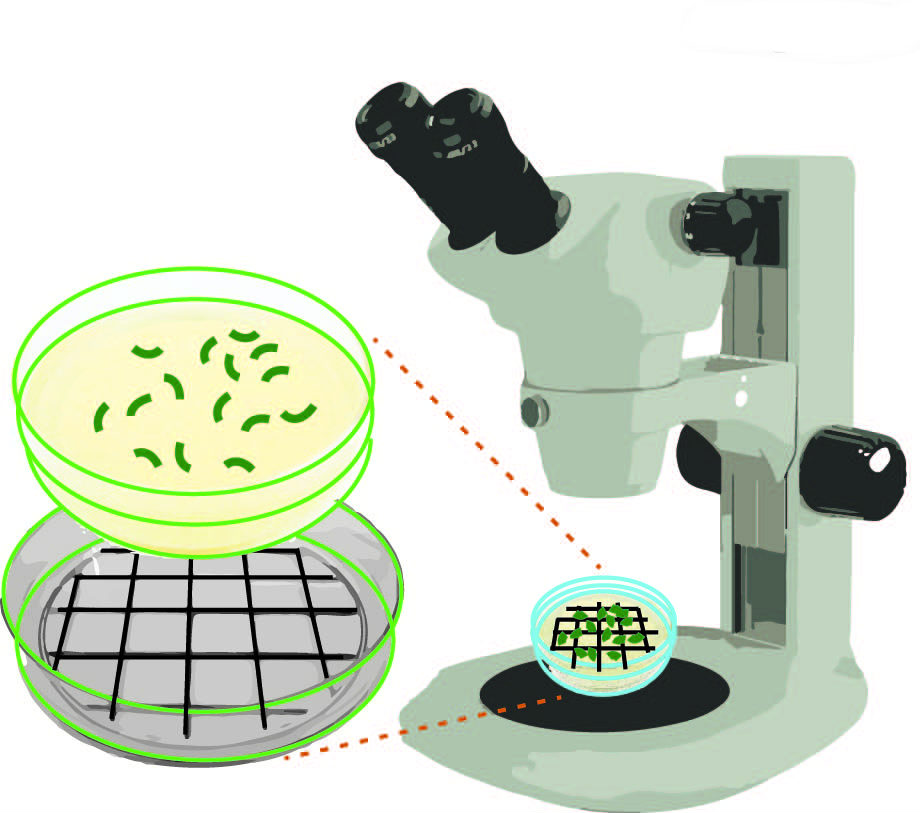
Figure 3. Schematic representation of how to score numbers of eggs laid and hatched on NGM agar plates. Using a dark colored marker draw a grid on a Petri dish lid. This lid can be placed under your plate to facilitate scoring under a stereomicroscope.
- After incubation at 20 °C for 20-24 h, count numbers of unhatched/dead eggs and hatched F1 worms. For counting larvae, it is easier to count when they become L2/L3, however unhatched eggs will be degraded and therefore more difficult to see and count if you wait longer.

If necessary, continue to count larvae and adults at Day 9. Represent the data as either embryonic viability or larval lethality as shown in Figure 4.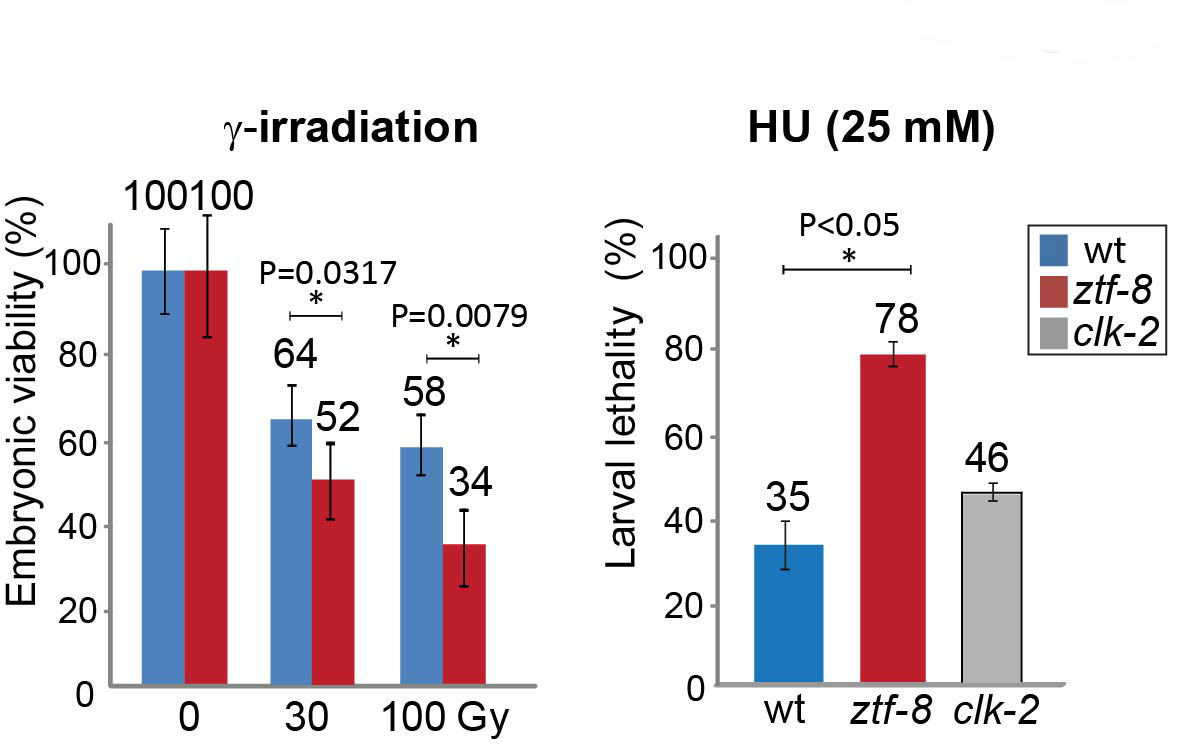
Figure 4. DNA damage sensitivity assay results for ztf-8 mutants. Representative data using graphs adapted from Kim and Colaiacovo (2014). Relative embryonic viability and larval lethality for ztf-8 and clk-2 mutants compared to wild type after treatment with the indicated doses of γ-IR and HU. The results from two independent experiments (biological replicates) are shown. Asterisks indicate statistical significance calculated by two-tailed Mann-Whitney test, 95% confidence interval. Error bars represent standard error of the mean.
- To prepare synchronized L1 stage larvae, gravid adult hermaphrodites are washed off from the plates by adding 1-2 ml of M9/plate. Leave the plates with M9 for 30-60 sec to dislodge the worms. Alternatively, for a small-scale test, L4 worms can be picked with a platinum wire instead of washing off the plate. However, synchronizing via bleaching removes potential fungal contamination of the worm which can be problematic when counting worms. The total number of adult worms required might vary, but 40 adults are required to obtain ~160 L1 larvae. If you have synchronized L4 worms ready for the assays, skip this step and start from step C10.
Notes
- Instead of spinning down worms using a centrifuge, worms can also be harvested by sedimentation at the bench for ~5 min.
- In general, over 25 animals were plated, 5 per plate. Each DNA damage-inducing condition was replicated at least twice in independent experiments.
- To avoid a thick bacterial lawn, seed 50 μl of overnight grown OP50 (OD600>2.0) liquid culture to NGM plates and spread it with a glass rod. Incubate 12-14 h at 37 °C.
Recipes
- 20% alkaline hypochlorite solution (45 ml)
Make fresh each time
24.75 ml ddH2O
11.25 ml 1 M NaOH
9.0 ml Bleach (non germicidal)
Acknowledgments
We thank Doris Lui, Elisabeth Altendorfer and Jinmin Gao for proof reading the manuscript. This work was supported by National Institutes of Health grants R01GM072551 and R01GM105853 to MPC. This protocol has been adapted from our previous work (Kim and Colaiacovo, 2014).
References
- Bailly, A. P., Freeman, A., Hall, J., Declais, A. C., Alpi, A., Lilley, D. M., Ahmed, S. and Gartner, A. (2010). The Caenorhabditis elegans homolog of Gen1/Yen1 resolvases links DNA damage signaling to DNA double-strand break repair. PLoS Genet 6(7): e1001025.
- Brenner, S. (1974). The genetics of Caenorhabditis elegans. Genetics 77(1): 71-94.
- He, F. (2011). Common worm media and buffers. Bio-protocol Bio101: e55.
- Jaramillo-Lambert, A., Harigaya, Y., Vitt, J., Villeneuve, A. and Engebrecht, J. (2010). Meiotic errors activate checkpoints that improve gamete quality without triggering apoptosis in male germ cells. Curr Biol 20(23): 2078-2089.
- Kessler, Z. and Yanowitz, J. (2014). Methodological considerations for mutagen exposure in C. elegans. Methods 68(3): 441-449.
- Kim, H. M. and Colaiacovo, M. P. (2014). ZTF-8 interacts with the 9-1-1 complex and is required for DNA damage response and double-strand break repair in the C. elegans germline. PLoS Genet 10(10): e1004723.
- Saito, T. T., Mohideen, F., Meyer, K., Harper, J. W. and Colaiacovo, M. P. (2012). SLX-1 is required for maintaining genomic integrity and promoting meiotic noncrossovers in the Caenorhabditis elegans germline. PLoS Genet 8(8): e1002888.
- Ward, J. D., Barber, L. J., Petalcorin, M. I., Yanowitz, J. and Boulton, S. J. (2007). Replication blocking lesions present a unique substrate for homologous recombination. EMBO J 26(14): 3384-3396.
Article Information
Copyright
© 2015 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Kim, H. and Colaiácovo, M. P. (2015). DNA Damage Sensitivity Assays in Caenorhabditis elegans. Bio-protocol 5(11): e1487. DOI: 10.21769/BioProtoc.1487.
Category
Molecular Biology > DNA > DNA damage and repair
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.