- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Determining the Relative Fitness Score of Mutant Viruses in a Population Using Illumina Paired-end Sequencing and Regression Analysis
Published: Vol 5, Iss 10, May 20, 2015 DOI: 10.21769/BioProtoc.1475 Views: 9412
Reviewed by: Arsalan DaudiMigla MiskinyteAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Apr 2014
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
Recent advances in DNA sequencing capacity to accurately quantify the copy number of individual variants in a large and diverse population allows in parallel determination of the phenotypic effects caused by each genetic modification. This systematic profiling approach is a combination of forward and reverse genetics, which we refer to as quantitative high-resolution genetics (qHRG). This protocol describes how to determine the relative fitness score of each variant compared to wild type (WT) virus based on its frequency determined by Illumina sequencing. Random mutagenesis techniques will be used to introduce randomization at each codon position of the targeted region, thereby generating a comprehensive input mutant library with substitutions at each position of interest (Qi et al., 2014; Wu et al., 2014a; Wu et al., 2014b). After selection, each selected library will be sequenced by Illumina paired-end sequencing and the frequency of each mutation will be determined. Based on the change in frequency, the relative fitness score of each mutant can be calculated with regression analysis.
Materials and Reagents
- The Huh-7.5.1 cell line (kindly provided by Dr. Francis Chisari from the Scripps Research Institute, USA)
- Dulbecco's modified Eagle medium (DMEM) (Corning, Cellgro®, catalog number: 10-017-CV )
- Fetal bovine serum (FBS) (Omega Scientific, catalog number: FB-11 )
- 100x non-essential amino acids solution (Life Technologies, catalog number: 11140050 )
- 1 M HEPES (Life Technologies, catalog number: 15630080 )
- 100x Penicillin-Streptomycin-Glutamine (Life Technologies, catalog number: 10378016 )
- 10x trypsin supplemented with EDTA (Life Technologies, Gibco®, catalog number: 15400054 )
- Plasmid that carries the HCV viral genome (pFNX-HCV) was synthesized based on the chimeric sequence of J6/JFH1 virus
Note: In this protocol, we are taking the HCV NS5A mutant library as an example to describe the procedures to relative fitness determination (Qi et al., 2014). A mutant virus library where each codon of interest was individually substituted with ‘NNK’, where N represents random incorporation of A/T/G/C; K represents random incorporation of T/G. The randomized codons therefore include 32 nucleotide combinations, which cover all possible amino acid. - 100% ethanol (Decon Labs, catalog number: 2701 )
- QIAamp Viral RNA Mini Kit for viral RNA purification (QIAGEN, catalog number: 52906 )
- Sterile, RNase-free pipet tips (with aerosol barriers for preventing cross-contamination) (OLYMPUS, catalog numbers: 24-401 , 24-404 , 24-412 , 24-430 )
- SuperScriptTM III Reverse Transcriptase Kit (Life Technologies, InvitrogenTM, catalog number: 18080-044 )
- RNaseOUT Recombinant Ribonuclease Inhibitor (Life Technologies, InvitrogenTM, catalog number: 10777-019 )
- KOD Hot Start DNA Polymerase Kit (Novagen®, catalog number: 71086-4 )
- PureLink® Quick PCR Purification Kit (Life Technologies, InvitrogenTM, catalog number: K3100-02 )
- T4 Polynucleotide Kinase (PNK) (New England Biolabs, catalog number: M0201S )
- NEB buffer 2 (New England Biolabs, catalog number: B7002S )
- dATP (100 mM) (Life Technologies, InvitrogenTM, catalog number: 10216-018 )
- Klenow Fragment (3’ to 5’ exo-) enzyme (New England Biolabs, catalog number: M0212S )
- T4 DNA Ligase Kit (Life Technologies, InvitrogenTM, catalog number: 15224-017 )
Equipment
- 15 cm cell culture dishes (Genesee Scientific, catalog number: 25-203 )
- T-150 cell culture flasks (Genesee Scientific, catalog number: 25-211 )
- 37 °C, 5% CO2 cell culture incubator
- 1.7 ml Microtubes (1.5 ml) (Genesee Scientific, catalog number: 22-282 )
- Falcon 50 ml tubes (Corning, catalog number: 14-432-22 )
- Falcon 15 ml tubes (Corning, catalog number: 05-527-90)
- Microcentrifuge (with rotor for 1.5 ml and 2 ml tubes) (Eppendorf, model: 5424 )
- Centrifuge (with rotor for 15 ml and 50 ml Falcon tubes) (Thermo Fisher Scientific, Legend RT)
- NanoDrop ND-1000 UV Spectrophotometer (Thermo Fisher Scientific)
- Thermal cycler (Eppendorf, catalog number: 950030050 )
Procedure
- Passage the mutant virus library (pool 1) in Huh-7.5.1 cells for selection
- Seed Huh-7.5.1 cells in T-150 cell culture flasks at 50% confluence (approximately 4 million cells in 24 ml of complete growth medium).
- Aspire growth medium in the flask using a Pasteur pipette and infect the monolayer cells with mutant HCV library at M.O.I = 0.1 [the virus library should be titrated in advance as described earlier by Arumugaswami et al. (2008)].
- Incubate the cells at 37 °C incubator for 6 h. Aspirate old medium and put 24 ml of fresh complete growth medium (DMEM with 10% of FBS, 1x NEAA and 1x Penicillin/Streptomycin/Glutamine).
- Incubate the virus infected cells for 72 h at 37 °C before Huh7.5.1 cells reach 100% confluence (approximately 8 million cells).
- Collect the supernatant in a 50 ml Falcon tube.
- Wash the cells with 1x PBS once.
- Trypsinize the cells with 2 ml of 1x trypsin for 1 min at RT and tap flask to completely loosen cells.
- Stop trypsin by adding 24 ml of complete growth medium as mentioned in step A3.
- Distribute cells to 3 new flasks at 8 ml/flask.
- Distribute 8 ml of collected supernatant from step A5 into each flask from step A9, and add 8 ml of fresh complete growth medium into each flask to reach 24 ml/flask.
- Incubate the virus infected cells for 72 h at 37 °C before they reach 100% confluence.
- Collect the supernatant (144 h post infection) and store as library pool 2.
- Titrate the virus titer in pool 2.
- Repeat steps from A1 to A13 to passage the pool 2 and collect pool 3.
- Repeat steps from A1 to A13 to passage the pool 3 and collect pool 4.
- Repeat steps from A1 to A13 to passage the pool 4 and collect pool 5.
- Seed Huh-7.5.1 cells in T-150 cell culture flasks at 50% confluence (approximately 4 million cells in 24 ml of complete growth medium).
- Determine the frequency of each mutant virus at each passage
- Extract HCV genomic RNA from each pool (pool 1 through pool 5) with QIAamp Viral RNA Mini Kit for viral RNA purification from QIAGEN. All of the reagents used in this step are all from this kit, if not otherwise stated.
- The supernatant of each virus pool was spun at 1,500 x g for 10 min to get rid of possible contamination from cell associated RNA.
- Take 1.4 ml of supernatant from each sample in a 15 ml Falcon tube.
- Lyse the virus with 5.6 ml of lysis buffer (AVL) containing 1 μg/ml of carrier RNA (5.6 μg of total carrier RNA per sample to avoid overload of the columns) by pulse-vortexing for 15 sec and incubate at room temperature for 10 min.
- Add 5.6 ml of ethanol (100%) to the sample, and mix by pulse-vortexing for 15 sec.
- Transfer 630 μl of the solution from step 4 to the QIAamp Mini column (in a 2 ml collection tube). Close the cap and centrifuge at 6,000 x g for 1 min and discard the filtrate collected in the collection tube.
- Repeat step 5 until all of lysate step 4 is loaded onto the spin column.
- Add 500 μl of buffer AW1 onto the QIAamp Mini column, and centrifuge at 6,000 x g for 1 min.
- Place the QIAamp Mini column in a clean 2 ml collection tube and discard the filtrate.
- Add 500 μl of buffer AW2 and centrifuge at 20,000 x g for 1 min. Discard the filtrate collected in the collection tube.
- Centrifuge at full speed (20,000 x g) for 2 min to completely dry the column.
- Place the QIAamp Mini column in a clean 1.5 ml Eppendorf tube and add 60 μl of buffer AVE to the filter area of the column. Close the cap and incubate at room temperature for 1 min. Spin at full speed (20,000 x g) for 1 min to elute the RNA.
- The supernatant of each virus pool was spun at 1,500 x g for 10 min to get rid of possible contamination from cell associated RNA.
- Reverse transcription reaction and PCR amplification of the targeted region for sequencing. We use SuperScriptTM III Reverse Transcriptase kit from Life Technologies, and all of the reagents are from the kit if not otherwise stated.
- Set up 20 μl reverse transcription reaction with 10 μl of RNA isolated from each pool (pool 1-5) and the input RNA library (pool 0) which was used to reconstitute the mutant virus library as mentioned by Qi et al. (2014). Add the following components to a nuclease-free Eppendorf tube:
RNA isolated from each pool 10 μl Random primer (100 ng/ul) 1 μl dNTP (10 mM) 1 μl H2O 1 μl Total 13 μl - Incubate the mixture at 65 °C for 5 min and incubate on ice for 1 min.
- Spin down the tube for 5 sec and add the following components:
RNA mixture from step 2 13 μl 5x First-Strand Buffer 4 μl 0.1 M DTT 1 μl RNaseOUT RNase inhibitor 1 μl SuperScript III RT 1 μl Total 20 μl - Incubate at 25 °C for 5 min and 50 °C for 60 min.
- Inactivate the reaction by heating at 70 °C for 15 min.
- Determine the virus genome copy number in each pool with Q-PCR using a pair of HCV-specific primer as follows (Arumugaswami et al., 2008):
Primer_forward: AGA GCC ATA GTG GTC TGC G
Primer_reverse: CTT TCG CAA CCC AAC GCT AC - Amplify the targeted region with PCR using KOD DNA polymerase for “just enough” cycle numbers (based on the Q-PCR reaction in step 2f) to reach saturation. For example, We would use 28 PCR amplification cycles at this step if 30 cycles would saturate the reaction according to the Q-PCR result.
- Purify the PCR amplicon from each PCR reaction with PCR purification kit from Life Technologies and measure the concentration of each sample with NanoDrop ND-1000 Spectrophotometer.
- Set up 20 μl reverse transcription reaction with 10 μl of RNA isolated from each pool (pool 1-5) and the input RNA library (pool 0) which was used to reconstitute the mutant virus library as mentioned by Qi et al. (2014). Add the following components to a nuclease-free Eppendorf tube:
- Construct sequencing samples for Illumina sequencing.
- Take 1 μg of each PCR amplicon product from each sample and set up the following reaction with T4 Polynucleotide Kinase (PNK) to add 5’-phosphate to amplicons to allow subsequent ligation.
PCR amplicons 5-17 μl (1 μg total) T4 PNK Reaction Buffer 2 μl T4 PNK 1 μl H2O 0-12 μl Total 20 μl - Incubate at 37 °C for 1 h and purify the sample with PCR purification columns in 40 μl.
- dA-Tailing with Klenow Fragment (3'-->5' exo-):
PCR amplicons 37 μl NEB buffer 2 (10x) 5 μl dATP (1 mM) 5 μl Klenow Fragment (3’ to 5’ exo-) 3 μl Total 50 μl - Incubate at 37 °C for 30 min and purify DNA samples with PCR purification columns in 35 μl volume.
- Ligate with Illumina sequencing adaptors with various barcodes designating to different pools:Adapters were generated by annealing two oligos:
PCR amplicons 30 μl T4 DNA ligase reaction buffer (10x) 5 μl Adaptor with barcodes (10uM) 5 μl T4 DNA ligase 2 μl Sterile H2O 8 μl Total 50 μl
5'-ACA CT CTT TCC CTA CAC GAC GCT CTT CCG ATC TNN NT-3' 5'-/5Phos/NNN AGA TCG GAA GAG CGG TTC AGC AGG AAT GCC GAG-3'. The location of multiplex ID for distinguishing different samples is underlined. NNN represents different sequences of multiplex ID. - Incubate at 25 °C (room temperature) for 1 h and purify with PCR purification columns in 30 μl volume.
- The adapter-ligated products were enriched by a final PCR using primers:
5'-AAT GAT ACG GCG ACC ACC GAG ATC TAC ACT CTT TCC CTA CAC GAC-3' 5'-CAA GCA GAA GAC GGC ATA CGA GAT CGG TCT CGG CAT TCC TGC TGA ACC-3'. - Purify the DNA with PCR purification columns in 30 μl volume and measure concentrations with NanoDrop ND-1000 Spectrophotometer.
- Mix 500 ng of final product from each pool and submit for Illumina sequencing (HiSeq).
- Take 1 μg of each PCR amplicon product from each sample and set up the following reaction with T4 Polynucleotide Kinase (PNK) to add 5’-phosphate to amplicons to allow subsequent ligation.
- Extract HCV genomic RNA from each pool (pool 1 through pool 5) with QIAamp Viral RNA Mini Kit for viral RNA purification from QIAGEN. All of the reagents used in this step are all from this kit, if not otherwise stated.
- Determine the frequency of each mutant virus at each passage and calculate relative fitness score of each mutant virus with regression analysis.
- Each pair-end sequence read in the HiSeq data file was mapped to the reference sequence once it passes the quality control (cut off 35). Each miss match from the reference sequence will be identified as a mutation and the number of each mutation will be counted. The script ‘mapping.txt’ for mutation mapping is provided here.
- Calculate the frequency of a given variant, v, in the pool #N (fv,N) and the frequency of WT, wt, in the pool #N (fwt,N) as follows:
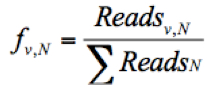
(The frequency of the given variant in pool #N)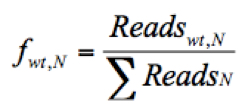
(The frequency of the WT virus in pool #N)
Where Readsv,N indicates the number of sequence reads for the variant (v) in pool #N, Readswt,N shows the number of sequence reads for the WT in pool #N, and ΣReadsN represents the total reads in the pool #N. - Discard any frequency that is lower than 0.0005, since the mutation frequency of HCV is about 10-5 to 10-4 nucleotide substitutions per nucleotide per round of genome replication.
- Calculate the relative fitness score of each mutant virus. The relative fitness score of a given variant (Wv) was determined as the antilogarithm of the slope of the regression using the following formula implemented in Python:
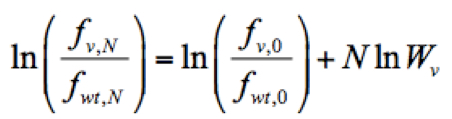
Where ln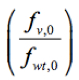 is the logarithm of the relative frequency of a given variant (v) in the input RNA library, pool 0, which was used to reconstitute the mutant virus library. Script ‘fitness_reg.txt’ for fitness calculation is provided here.
is the logarithm of the relative frequency of a given variant (v) in the input RNA library, pool 0, which was used to reconstitute the mutant virus library. Script ‘fitness_reg.txt’ for fitness calculation is provided here.
- Each pair-end sequence read in the HiSeq data file was mapped to the reference sequence once it passes the quality control (cut off 35). Each miss match from the reference sequence will be identified as a mutation and the number of each mutation will be counted. The script ‘mapping.txt’ for mutation mapping is provided here.
Representative data
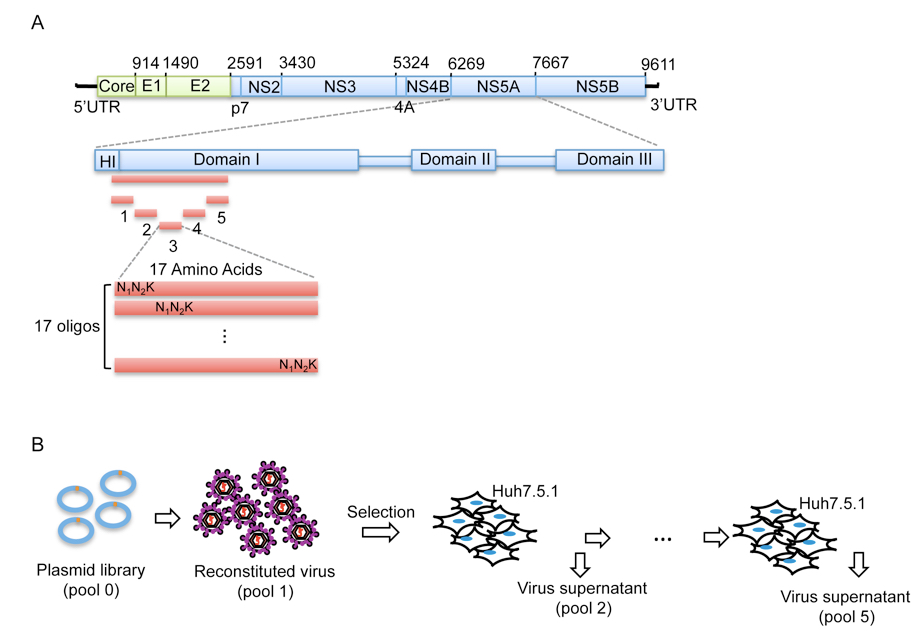
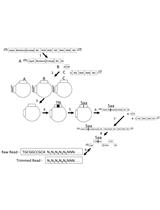
Figure 1. Procedure of mutant library construction and selection. A. Schematic picture showing the construction of the saturation mutant library in a sub-domain of NS5A of HCV. The area to be mutated was divided into 5 small regions, and each of them was composed of 17 or 18 amino acids. Each residue was replaced with one random codon (N1N2K: N1 and N2 codes for A/T/G/C and K codes for T/G) and incorporated into the WT background of HCV. B. The resultant viral library was then selected in vitro by passing through Huh5.7.1 cells for 4 rounds.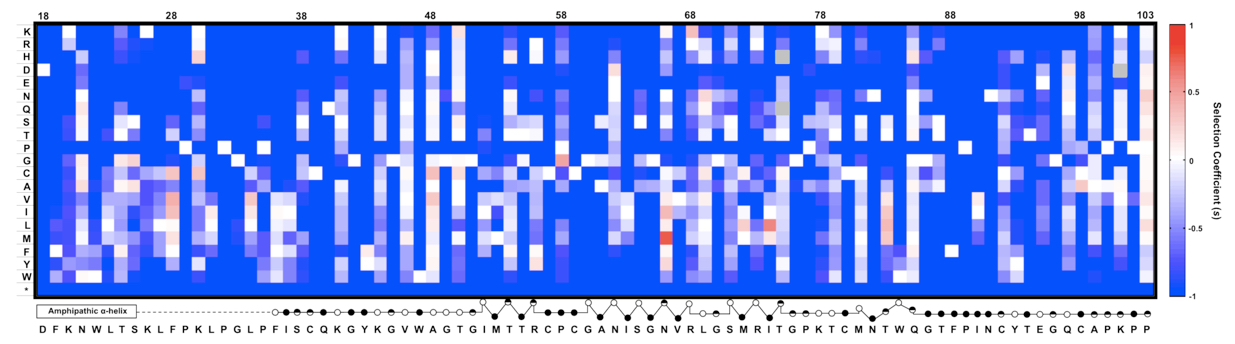
Figure 2. An example of expected data: The fitness landscape of amino acids 18-103 in NS5A in virus replication. This is a heat map showing the relative fitness scores represented as selection coefficient (s) for each variant during viral replication in vitro. Color indicates the fitness of each mutant calculated as ‘s’ relative to WT (Materials and method). Red represents positive ‘s’ (i.e. increased fitness) and blue represents negative ‘s’. s = 0 means the same fitness as the WT virus. The secondary structure of the mutated region is annotated below the figure (open circles: solvent exposed residues; filled circles: buried residues; half-filled circles: partially buried residue). This figure was generated by MATLAB software.
Notes
- During the process of passaging the mutant virus library in Huh-7.5.1 cells for in vitro selection, the library complexity should be estimated and always be maintained throughout the entire procedure. The complexity of library can be estimated depending on the way of the library is constructed. For example, in our recent study by Qi et al. (2014), we substituted each of the 86 position in the region of NS5A (from a.a. 18 to a.a. 103) with all possible 20 amino acids plus stop codon. In this case, the library complexity can be calculated as: 86 x 20 (19 variants plus stop codon) + 1 (WT) = 1721. According to our experience, we found that covering each variant for at least 100x on average gives optimal and reproducible results.
- The library should be selected for multiple rounds for regression analysis to give much higher confidence when calculating the relative fitness scores.
Acknowledgments
This work was supported by the following grants: National Natural Science Foundation of China (NSFC) 81172314, National Science Foundation EF-0928690 (JLS) and National Institute of Health AI078133 (RS), Margaret E. Early Medical Research Trust, P30CA016042 (Jonson Comprehensive Cancer Center) and P30AI028697 (UCLA AIDS Institute/CFAR). JLS is grateful for the support of the De Logi Chair in Biological Sciences and the RAPIDD program of the Science & Technology Directorate of the US Department of Homeland Security, and the Fogarty International Center, National Institutes of Health. C.A.O. was supported by the NCI Cancer Education Grant, R25 CA 098010.
References
- Qi, H., Olson, C. A., Wu, N. C., Ke, R., Loverdo, C., Chu, V., Truong, S., Remenyi, R., Chen, Z., Du, Y., Su, S. Y., Al-Mawsawi, L. Q., Wu, T. T., Chen, S. H., Lin, C. Y., Zhong, W., Lloyd-Smith, J. O. and Sun, R. (2014). A quantitative high-resolution genetic profile rapidly identifies sequence determinants of hepatitis C viral fitness and drug sensitivity. PLoS Pathog 10(4): e1004064.
- Wu, N. C., Young, A. P., Al-Mawsawi, L. Q., Olson, C. A., Feng, J., Qi, H., Luan, H. H., Li, X., Wu, T. T. and Sun, R. (2014). High-throughput identification of loss-of-function mutations for anti-interferon activity in the influenza A virus NS segment. J Virol 88(17): 10157-10164.
- Wu, N. C., Young, A. P., Al-Mawsawi, L. Q., Olson, C. A., Feng, J., Qi, H., Chen, S. H., Lu, I. H., Lin, C. Y., Chin, R. G., Luan, H. H., Nguyen, N., Nelson, S. F., Li, X., Wu, T. T. and Sun, R. (2014). High-throughput profiling of influenza A virus hemagglutinin gene at single-nucleotide resolution. Sci Rep 4: 4942.
- Arumugaswami, V., Remenyi, R., Kanagavel, V., Sue, E. Y., Ngoc Ho, T., Liu, C., Fontanes, V., Dasgupta, A. and Sun, R. (2008). High-resolution functional profiling of hepatitis C virus genome. PLoS Pathog 4(10): e1000182.
Article Information
Copyright
© 2015 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Qi, H., Olson, C. A., Wu, N. C., Du, Y. and Sun, R. (2015). Determining the Relative Fitness Score of Mutant Viruses in a Population Using Illumina Paired-end Sequencing and Regression Analysis . Bio-protocol 5(10): e1475. DOI: 10.21769/BioProtoc.1475.
Category
Microbiology > Microbial genetics > Mutagenesis
Systems Biology > Genomics > Sequencing
Molecular Biology > RNA > RNA sequencing
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.