- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Whole Genome Bisulfite Sequencing and DNA Methylation Analysis from Plant Tissue

Published: Vol 5, Iss 4, Feb 20, 2015 DOI: 10.21769/BioProtoc.1407 Views: 21197
Reviewed by: Tie LiuEmily CopeAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Jul 2014
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
This protocol describes whole genome bisulfite-sequencing library preparation from plant tissue and subsequent data analysis. Allele-specific methylation analysis and genome-wide identification of differentially methylated regions are additional features of the analysis procedure.
Part I. Whole genome bisulfite sequencing library preparation
Materials and Reagents
- RNAse-treated DNA (at least 1 µg, volume should not exceed 130 µl)
- AMPure XP beads (Beckman Coulter, catalog number: A63880 )
- 200 proof Ethyl alcohol (Sigma-Aldrich, catalog number: 459844-500ML )
- Illumina TruSeq kit (Illumina, catalog number: FC-121-2001 )
- Methylcode Invitrogen kit (Life Technologies, InvitrogenTM, catalog number: MECOV50 )
Note: Other bisulfite conversion kits can be used, but were not tested in this protocol. - T4 DNA ligase (NEB, catalog number: M0202T )
- Pfu Cx Hotstart DNA polymerase (Agilent, catalog number: 600412 )
- TOPO Blunt cloning kit (Life Technologies, InvitrogenTM, catalog number: 450245 )
- TOPO TA cloning kit (Life Technologies, InvitrogenTM, catalog number: 450030 )
- Ex Taq DNA polymerase (TaKaRa, Clontech, catalog number: RR001B )
- 1 Kb-plus DNA ladder (Life Technologies, InvitrogenTM, catalog number: 10787-018 )
- Agarose
- 1x TAE buffer (see Recipes)
Equipment
- Agarose gel electrophoresis apparatus
- Covaris microTUBE prep station (Covaris, part number: 500142 )
- Covaris microTUBE AFA Fiber Pre-Slit Snap-Cap (6 x 16 mm) (Covaris, part number: 520045 )
- Low-retention 1.5 ml tubes (Thermo Fisher Scientific, catalog number: 02-681-320 )
- TempAssure PCR 8-Strips (USA Scientific, catalog number: 1402-4700 )
- Magnetic stand for 1.5 ml tubes (DynaMagTM-2 magnet, Life Technologies, InvitrogenTM, catalog number: 12321D )
- Focused-ultrasonicator (Covaris, S-Series, model: S220 )
- Microcentrifuge (VWR International, catalog number: 93000-204 )
- Thermocycler
Procedure
- Prepare reagents before starting
- Make fresh 80% ethanol.
Tip: It is important to use fresh 80% ethanol for Ampure bead purification. - Shake the Agencourt AMPure XP bottle to resuspend beads. 180 µl of beads will be needed per sample (ratio of 1.4:1 beads to DNA). Keep at room temperature.
- Make fresh 80% ethanol.
- Shear DNA with Covaris sonicator
- Fill tank with deionized water to 12.5 on the graduate fill line label.
- Turn on the machine, the chiller (set temperature at +3 °C), and the computer.
- Launch the application software (Covaris SonoLab 7). Push the degas button. Degas the instrument for at least 30 min before use.
- Settings: Peak power 175 W, duty factor 10, cycles/burst 200, time 6 min, temperature 6 °C. Note that the temperature rises while the machine is in use, which could interfere with the shearing cycle. Thus, the machine is initially set to 3 °C as a precaution.
- Transfer 130 µl of DNA sample (~10 ng/µl) into the Covaris microTUBE, spin down briefly and proceed with shearing.
- Run 1.2% gel with sheared DNA (250 ng) and un-sheared DNA side by side to evaluate shearing. Samples can also be run on a Bioanalyzer. Expected size range is 100-300 bp. See representative data Figure 1.
- Fill tank with deionized water to 12.5 on the graduate fill line label.
- DNA purification (AMPure beads)
Refer to Agencourt AMPure XP PCR Purification - Instructions For Use (B37419AA)
(https://www.beckmancoulter.com/wsrportal/techdocs?docname=B37419AA).- Place the tube into Covaris microTUBE Prep Station and remove the cap. Transfer the 130 µl of sample to a low-DNA binding microfuge tube. Add 180 µl of resuspended beads and mix well by pipetting.
- Incubate for 5 min at room temperature.
- Place the tube onto the magnetic stand for 2 min to separate beads from the solution.
- Aspirate the cleared solution from the tube and discard. Do not disturb the beads.
- Dispense 250 μl of 80% ethanol to the tube and incubate for 30 sec at room temperature.
- Aspirate the ethanol and discard. Repeat for a total of two washes.
- Let the beads air dry for 5 min.
Tip: Be careful not to over-dry the beads (bead pellet appears cracked if over-dried) as this will significantly decrease elution efficiency. - Remove the microfuge tube from the magnetic stand. Add 52 μl of water to the beads and resuspend the beads by pipetting 10 times. Incubate for 2 min at room temperature.
- Place the tube back on the magnetic stand. Transfer 50 µl of the clear supernatant to a new PCR tube without disturbing the beads.
- Place the tube into Covaris microTUBE Prep Station and remove the cap. Transfer the 130 µl of sample to a low-DNA binding microfuge tube. Add 180 µl of resuspended beads and mix well by pipetting.
- End Repair of the purified sheared DNA (Illumina TruSeq kit)
- Preheat thermocycler to 30 °C.
- Add 10 µl of Resuspension Buffer and 40 µl End Repair Mix to the sheared DNA from step C9.
- Adjust pipette to 50 µl then gently pipette the entire volume up and down 10 times.
- Incubate for 30 min at 30 °C.
- Preheat thermocycler to 30 °C.
- DNA Purification (AMPure beads)
- Vortex the AMPure beads until they are well dispersed.
- Prepare a diluted bead mixture by combining 125 µl beads with 35 µl of PCR grade water.
- Add the entire volume from step D4 (100 µl) to 160 µl of the diluted beads.
- Adjust pipette to 200 µl then gently pipette the entire volume up and down 10 times.
- Incubate at RT for 10 min.
- Place the tube on the magnetic stand for 5 min.
- Remove and discard the supernatant from each tube without disturbing the beads.
- With the tube on the magnetic stand, add 200 µl of freshly prepared 80% ethanol without disturbing the beads. Incubate 30 sec.
- Remove and discard all the supernatant from each tube without disturbing the beads. Repeat ethanol wash once.
- Let the tube stand at RT to dry for 5-8 min. Remove the tube from the stand and resuspend the dried pellet with 17.5 µl Resuspension Buffer from the Illumina TruSeq kit. Gently pipette the entire volume up and down 10 times to mix thoroughly.
- Incubate the tube at RT for 2 min.
- Place the tube on the magnetic stand at RT for 3 min.
- Transfer 15 µl of the clear supernatant to a new PCR tube without disturbing the beads.
SAFE STOPPING POINT DNA can be stored at -20 °C for up to 7 days.
- Vortex the AMPure beads until they are well dispersed.
- Adenylate 3’ Ends (Illumina TruSeq kit)
- Remove the A tailing mix from -20 °C and thaw at RT.
- Preheat thermal cycler at 37 °C.
- Add 2.5 µl of Resuspension buffer and 12.5 µl of A tailing mix to the tube from step E13.
- Adjust pipette to 30 µl, then gently pipette the entire volume up and down 10 times.
- Incubate at 37 °C for 30 min.
- Immediately proceed to adapter ligation.
- Remove the A tailing mix from -20 °C and thaw at RT.
- Ligate Adapters (Illumina TruSeq kit)
Illumina TruSeq DNA adapters, which contain 5-methylcytosines instead of cytosines, are ligated in a 50 μl reaction.- Remove the Adapter Index tubes from -20 °C and thaw at RT.
- Combine sample and reagents as indicated.
DNA sample (from step F6) 30 µl Adapters 2.5 µl T4 DNA Ligase (2,000,000 U/ml) 2.5 µl 10x T4 Ligase buffer with ATP 5 µl Water 10 µl - Incubate overnight at 16 °C.
- Remove the Adapter Index tubes from -20 °C and thaw at RT.
- DNA Purification (AMPure beads)
- Vortex the AMPure beads until they are well dispersed.
- Add 42.5 µl of the beads to the sample.
- Adjust pipette to 85 µl then gently pipette the entire volume up and down 10 times.
- Incubate at RT for 10 min.
- Place the tube on the magnetic stand for 5 min.
- Remove and discard 80 µl of supernatant from each tube.
- With the tube on the magnetic stand, add 200 µl of freshly prepared 80% ethanol without disturbing the beads. Incubate 30 sec.
- Remove and discard all the supernatant from each tube without disturbing the beads. Repeat ethanol wash once.
- Let the tube stand at RT to dry for 8 min. Remove the tube from the stand and resuspend the dried pellet with 22.5 µl Resuspension Buffer. Gently pipette the entire volume up and down 10 times to mix thoroughly. Incubate at RT for 2 min.
- Place the tube on the magnetic stand at RT for 5 min.
- Transfer 20 of µl of the clear supernatant to new tube without disturbing the beads.
SAFE STOPPING POINT. DNA can be stored at -20 °C for up to 7 days.
- Vortex the AMPure beads until they are well dispersed.
- Bisulfite treatment
- Use the MethylCode™ Bisulfite Conversion Kit Invitrogen following the manufacturer’s protocol.
http://tools.lifetechnologies.com/content/sfs/manuals/methylcode_bisulfite_man.pdf - Elute bisulfite-treated DNA in 10 µl.
- Use the MethylCode™ Bisulfite Conversion Kit Invitrogen following the manufacturer’s protocol.
- Library enrichment PCR
Use 3 µl (from step I2) as a template in each of two PCR reactions.
Thermal cycler program:
95 °C for 2 min
12-15 cycles*:
95 °C for 20 sec
60 °C for 30 sec
72 °C for 1 min
72 °C for 7 min
*Keep the number of cycles as low as possible to reduce PCR duplicates.
PCR master mix (50 µl reaction)10x Pfu polymerase buffer 5 µl 10 mM dNTPs 1 µl PCR primer cocktail (Illumina TruSeq kit) 5 µl Pfu Cx Hotstart DNA polymerase 1 µl Water 35 µl - Library purification
- Make fresh 80% ethanol.
- Vortex the AMPure beads until they are well dispersed.
- Add 50 µl of beads to the sample.
- Adjust pipette to 85 µl then gently pipette the entire volume up and down 10 times.
- Incubate at RT for 10 min.
- Place the tube on the magnetic stand for 5 min.
- Remove and discard 80 µl of supernatant from each tube.
- With the tube on the magnetic stand, add 200 µl of freshly prepared 80% ethanol without disturbing the beads. Incubate 30 sec.
- Remove and discard all the supernatant from each tube without disturbing the beads. Repeat ethanol wash once.
- Let the tube stand at RT to dry for 8 min. Remove the tube from the stand and resuspend the dried pellet with 16 µl water. Gently pipette the entire volume up and down 10 times to mix thoroughly. Incubate at RT for 2 min.
- Place the tube on the magnetic stand at RT for 5 min.
- Transfer 15 of µl of the clear supernatant to new tube without disturbing the beads.
SAFE STOPPING POINT. DNA can be stored at -20 °C for up to 7 days.
- Make fresh 80% ethanol.
- Library validation
- Subject libraries to quality control on a Bioanalyzer before sequencing. Libraries should have a size range between 250 and 400 bp and the adapter dimer peak, if present, should be less than 10% of the library. See Figure 2 in representative data.
- Clone 1-2 µl into TOPO Blunt and sequence a few clones to check that the adapters are ligated as expected.
- Bisulfite conversion checkpoint: In plants, the chloroplast genome is expected to be unmethylated. Amplify chloroplast DNA from the library by PCR. Clone into TOPO TA cloning kit following manufacturer’s protocol and sequence by standard Sanger sequencing. Each C in the amplified PCR product should be converted and sequenced as a T.
Template: 2 µl libraryOligos for chloroplast DNA (Groszmann et al., 2011):10x Ex Taq buffer (Mg2+ plus) 5 µl 10 mM dNTPs 1 µl 10 µM For Oligo 2.5 µl 10 µM Rev Oligo 2.5 µl TaKaRA Ex taq (5 units/µl) 0.5 µl Water 36.5 µl
For: 5’-ATGATGTTGTTAGAATTTYATATAGG-3’
Rev: 5’-CATCATTTARCTATCRCAATTCTTT-3’
Thermal cycler program:
95 °C for 2 min
40 cycles:
95 °C 15 sec
52 °C 30 sec
72 °C 2 min
72 °C 10 min
- Subject libraries to quality control on a Bioanalyzer before sequencing. Libraries should have a size range between 250 and 400 bp and the adapter dimer peak, if present, should be less than 10% of the library. See Figure 2 in representative data.
- High throughput sequencing
Sequence library (~10 pM) on Illumina HiSeq 2500 machine. Paired end or 80 bp single end reads allow for better mapping to the genome, but standard 40 bp single end reads are also acceptable. Sequencing depth needed depends on the size of the genome and can be calculated using the Lander/Waterman equation.
The general equation is:
C = LN / G
• C stands for coverage
• G is the haploid genome length
• L is the read length
• N is the number of reads
Please refer to http://support.illumina.com/downloads/sequencing_coverage_calculator.html for more details.
Part II. Whole genome bisulfite sequencing data analysis
Equipment
- Linux computer with standard utility applications (including Perl)
R (http://www.r-project.org/; base installation only is needed)
Bismark (http://www.bioinformatics.babraham.ac.uk/projects/bismark/)
FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/)
FASTX Toolkit (http://hannonlab.cshl.edu/fastx_toolkit/)
SAMtools (http://samtools.sourceforge.net/)
bedtools (https://github.com/arq5x/bedtools2)
Procedure
Note: For all the scripts used in the Procedure, please download here.
- Quality control of sequenced reads
- Copy sequencing reads to desired location on local computer or server.
- Unzip and untar sequence files if needed.
gunzip file_name.txt.tar.gz
tar xvfp file_name.txt.tar - Run quality control of sequencing reads with fastqc (http://hannonlab.cshl.edu/fastx_toolkit/).
Usage: fastqc file_name.txt - Examine the fastqc report to determine if/how to filter and/or trim reads. Discard adapters and low quality reads (less than 75% quality scores above 25) if necessary.
Usage: fastq_quality_filter –q25 –p 75 –i file_name. txt –o file_name_trimmed.fastq - Re-run quality control to check final quality of the reads.
Usage: fastqc file_name_trimmed.fastq
- Copy sequencing reads to desired location on local computer or server.
- Align bisulfite reads with Bismark
Align the reads to the reference genome of your choice using Bismark (Krueger and Andrews, 2011).
We strongly encourage the reader to consult the review written by the authors of Bismark (http://www.ncbi.nlm.nih.gov/pubmed/22290186) before starting the data analysis. Here, we describe alignment of single end reads, but alignment of paired end reads is also possible.- Prepare bisulfite-treated genome for future mapping (only needs to be done once).
Usage: bismark_genome_preparation --path_to_bowtie reference_genome/fasta/ - Map BS-reads to converted genome (map forward and reverse reads separately).
Usage: bismark --non_directional -o bismark_file_name_trimmed.n1l50 -n 1 -l 50 reference_genome file_name.trimmed.fastq
Options are as follows:
-non_directional: reads are not strand-specific (a strand-specific option is also available)
-o output directory
-n max number of mismatches
-l length of seed (first number of nt that are mapped with <n mismatches)
*fastq: short reads in fastq format
Option #1: Libraries from a single inbred strain - Prepare bisulfite-treated genome for future mapping (only needs to be done once).
- Convert Bismark aligned reads from SAM to BAM, sort, and index
cd bismark_file_name_trimmed.n1l50
Usage: SAM_to_BAM_sort_index.pl file_name.fastq_bismark.sam
Expected output file: file_name.fastq_bismark.sorted.bam - Convert sorted BAM file to SAM format
Usage: samtools view -h file_name.fastq_bismark.sorted.bam > file_name_sorted.sam Samtools (Li et al., 2009) - Eliminate redundant reads
Using a sorted SAM file, keep only one sequence per strand that maps to the same start and end position. This eliminates PCR duplicates. A log file (SAM_redundancy_stats.log.txt) of counts for each read is created.
Usage: ./make_sorted_SAM_non-redundant_fewest_MM.pl file_name_sorted.sam > file_name_sorted_nr.sam
This is how it works:
The script reads a sorted SAM file and gets all the reads that map to the same position and strand and then- Sorts them by decreasing prevalence and then by increasing number of mismatches (not counting bisulfite conversions) to the genome.
- The most prevalent read with the total highest quality string is kept.
- In the case of a tie, the read with the fewest number of mismatches is retained.
- In the case of another tie, it prints out every unique read.
The output SAM file also has 3 new fields added on the end:
CT == count = number of times this read was found in the input file
MM == mismatches = number of mismatches compared to the genome sequence (not counting conversions)
QS == quality score = total quality score for this read
- Sorts them by decreasing prevalence and then by increasing number of mismatches (not counting bisulfite conversions) to the genome.
- Calculate a methylation value for each cytosine
Run Bismark’s supplementary script “bismark_methylation_extractor” on sorted SAM files.
Usage: bismark_methylation_extractor –s file_name_sorted_nr.sam -o bismark_output
This script creates 12 output files with methylation status of each C in three different contexts (CpG, CHG, CHH) with 4 mappings:
OT - original top strand
CTOT - complementary to original top strand
OB - original bottom strand
CTOB - complementary to original bottom strand
Shorthand for methylation call according to context:
z: unmethylated C in CpG context
Z: methylated C in CpG context
x: unmethylated C in CHG context
X: methylated C in CHG context
h: unmethylated C in CHH context
H: methylated C in CHH context - Calculate bisulfite conversion efficiency
The mean bisulfite conversion rate for each library is calculated based on the methylation status of each cytosine from reads mapping to the chloroplast genome, which is expected to be unmethylated.- Run Usage: ./methylation_summarizer_1.pl bismark_output > bismark_SampleName.summary_step1.txt
- Get info about methylation status of each nucleotide from each read mapping to the chloroplast.
Usage: grep ChrC bismark_*_trim3.n1l50.summary_step_1.txt > bismark_*_trim3.n1l50.summary_step_1.chloroplast.txt./methylation_summarizer_2_conversion.pl
bismark_*_trim3.n1l50.summary_step_1.chloroplast.txt >bismark_*_trim3.n1l50.conversion.txt
The output file contains the conversion at each position, and the mean C conversion across the chloroplast is printed to the screen.
Option #2: Libraries from F1 crosses - Run Usage: ./methylation_summarizer_1.pl bismark_output > bismark_SampleName.summary_step1.txt
- Map reads from hybrid libraries
In the case of F1 hybrid libraries, two parental genomes may be available as reference. You may decide to map reads to both the parental genomes to maximize the number of mapped reads using Bismark as described above. To do that, first map all the reads to the best reference genome, then align the “unmapped” reads to the other genome.
Make file of unmapped reads from the fastq file (e.g., reads not in SAM file).
Usage: ./get_fastq_reads_not_in_SAM.pl fastqFile samFile > reads.unmapped.fastq
To assign reads to a particular strain and to retain as many unique reads as possible, separate the reads by strand.
Script: split_bismark_SAM_by_read_strand.pl
Usage: split_bismark_SAM_by_read_strand.pl file_name_bismark.sam - Classify reads by parent-of-origin (allele specific DNA methylation analysis)
Classify the forward reads according to their parent-of-origin with a bedfile where the C>T SNPs between the two genomes of interest are ignored but all other SNPs are retained, and classify the reverse reads with a bedfile where the G>A SNPs between the two genomes of interest are ignored but all other SNPs are retained. There is only one bedfile, but read strand has to be specified (pos or neg).
Script: split_bismark_SAM_by_read_strand.pl
Usage: split_bismark_SAM_by_read_strand.pl file_name_bismark.sam
Expected output files: file_name_bismark.pos.sam, file_name_bismark.neg.sam
Script: classify_bismark_reads_by_parent.one_strand.pl
Usage: classify_bismark_reads_by_parent.one_strand.pl file_name_bismark.pos.sam SNPs.bed pos > file_name_bismark.pos_class.sam"
Usage: classify_bismark_reads_by_parent.one_strand.pl file_name_bismark.neg.sam SNPs.bed neg > file_name_bismark.neg_class.sam"
Reads are classified based on their sequence at known SNP positions. After classification, redundant reads from each class are eliminated.
To differentiate between 4 cases:- ST:Z: maternal
- ST:Z: paternal
- ST:Z: NE means no evidence for either genome
- ST:Z: both means evidence for both (conflicting data)
awk -F"\t" '$19 == "ST:Z: maternal " {print $0}' file_name_bismark.pos_class.sam > maternal.sam
awk -F"\t" '$19 == "ST:Z: paternal " {print $0}' file_name_bismark.pos_class.sam > paternal.sam
awk -F"\t" '$19 == "ST:Z:NE" {print $0}' file_name_bismark.pos_class.sam > NE.sam
awk -F"\t" '$19 == "ST:Z:both" {print $0}' file_name_bismark.pos_class.sam > both.sam
Run similar commands with file_name_bismark.neg_class.sam
- ST:Z: maternal
- Combine the SAM files
Example: cat file_name_neg.sam file_name_pos.sam > file_name_pos_neg.sam
You may delete the individual pos and neg files after checking the size of the combined files. - Prefix the header to the sam file
cat header file_name_pos_neg.sam > file_name_pos_neg.header.sam - Convert SAM to BAM, sort, and index
SAM_to_BAM_sort_index.pl file_name_pos_neg.header.sam
Expected outputfile: file_name_pos_neg.sorted.bam - Convert sorted BAM file to SAM format
samtools view -h file_name_pos_neg.sorted.bam > file_name_pos_neg.sorted.sam - Eliminate redundant reads (see step 5)
Script: make_sorted_SAM_non-redundant_fewest_MM.pl
Usage: make_sorted_SAM_non-redundant_fewest_MM.pl file_name_pos_neg.sorted.sam > file_name_pos_neg.sorted_nr.sam - Calculate a methylation value for each cytosine (see step 6)
Run methylation extractor for each set of classified reads as well as for all reads combined.
Usage: methylation_extractor -s file_name_pos_neg.sorted_nr.sam
Combine all the nr.sam files into an “allreads_nr.sam” file with cat command and run methylation_extractor again. - Summarize the methylation status across genomic windows
For each class, organize the 12 methylation extractor files in the output files folder. Divide the genome into 300 nt (windowWidth) windows, overlapping by 100 nt (windowOverlap).
Script: make_genome_windows_bed.pl (requiring a file of each chromosome and its length)
Usage: make_genome_windows_bed.pl chromInfo.txt windowWidth windowOverlap > genome_300nt_100_windows.bed
Script: Summarize_by_window.sh
Usage: bash Summarize_by_window.sh methylation_outputfiles genome_300nt_100_windows.bed
Summarize_by_window.sh features: This script summarizes methylation status across overlapping genomic windows of defined size by converting the processed Bismark methylation extractor output files into a set of bed files and determining weighted methylation values as described in Schultz et al. (2012). Bedgraph files for viewing in a genome browser are created in the bedgraph folder. Bismark's methylation extractor output by chr position (after sorting) is summarized by converting the methylation string into ummethylated counts, methylated counts, and percent methylation, producing output in BED-like format (scorePerPos folder). The folder weighted_summaries_by_window has three bed files, which merge sites across each window. For each window it provides counts and weighted percent methylation. - Identify differentially methylated regions (DMRs)
At least 5-read coverage at each site is required. Differential methylation is assayed by calculating the difference between samples (sample A-sample B of weighted methylation fractions), and confidence (p-value from Fisher's exact test) for each window in all sequence contexts is assigned. P values are corrected with the Benjamini and Hochberg False Discovery Rate (FDR). CG and CHG DMRs were defined as regions with a minimum overlap of 3 informative Cs between windows and, for example, a weighted methylation difference of at least 35 and a corrected p value < 0.01. CHH DMRs were defined as windows with a minimum 10 overlapping informative cytosines and, for example, a weighted methylation difference of at least 10 and a p value < 0.01. The user can use their own criteria for defining DMRs.
To compare mC window counts in two samples the following scripts are needed:
merge_bedgraph_data_counts_etc.pl
compare_methylation_counts_by_window.R- Make matrix of counts for 2 samples, each in 2 contexts using files in the 'weighted_summaries_by_window' folders.
- Example:
./merge_bedgraph_data_counts_etc.pl genome_300nt_100_windows.bed
sample_A_reads/weighted_summaries_by_window/CpG.bed
sample_B_reads/weighted_summaries_by_window/CpG.bed
sample_A_reads/weighted_summaries_by_window/CHG.bed
sample_B_reads/weighted_summaries_by_window/CHG.bed
sample_A_reads/weighted_summaries_by_window/CHH.bed
sample_B_reads/weighted_summaries_by_window/CHH.bed >
sample_A_vs_sample_B_reads.300nt_100_windows.txt
The resulting file should have window counts in this order:
sample_A/weighted_summaries_by_window/CpG.bed
sample_B/weighted_summaries_by_window/CpG.bed
sample_A/weighted_summaries_by_window/CHG.bed
sample_B/weighted_summaries_by_window/CHG.bed
sample_A/weighted_summaries_by_window/CHH.bed
sample_B/weighted_summaries_by_window/CHH.bed - Compare counts across two samples in the same context.
compare_methylation_counts_by_window.R inputCountsFile outputStatsFile
Usage:
compare_methylation_counts_by_window.R
sample_A_vs_sample_B_reads.300nt_100_windows.txt
sample_A_vs_sample_B_reads.300nt_100_windows.stats.txt"
For each window, the output file will have (a) the raw Fisher's exact test p-value reflecting whether the fraction of meth/unmeth counts is the same for both samples, and (b) the difference (sample A-sample B) in weighted methylation fraction. Methylation fractions appear next to the results of each statistical calculation.
- Make matrix of counts for 2 samples, each in 2 contexts using files in the 'weighted_summaries_by_window' folders.
- Overlap genomic features (genes, transposable elements, etc.) with the windows
- Open the stats file in Excel and sort based on p value.
- Copy the rows representing selected DMRs [(with a FDR below your threshold (0.01) and
with a methylation difference above your threshold (e.g. 35%)] into another file and delete all but the desired 5 columns (chr, start, end, difference, FDR value). - Save the file with the extension “bed” and intersect with genomic features using Bedtools (Quinlan and Hall, 2010).
Run:
bedtools intersect -wao -a sample_A_vs_sample_B.CpG.bed -b genome_GFF3_genes.gff > intersect_ sample_A_vs_sample_B_genes.txt
bedtools intersect -wao -a sample_A_vs_sample_B.CpG.bed –b genome_GFF3_transposable_element.gff > intersect_ sample_A_vs_sample_B.CpG.bed _TE.txt
- Open the stats file in Excel and sort based on p value.
Representative data
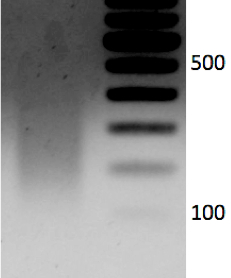
Figure 1. 1.2% gel with sheared DNA (250 ng). Expected size range is 100-300 bp.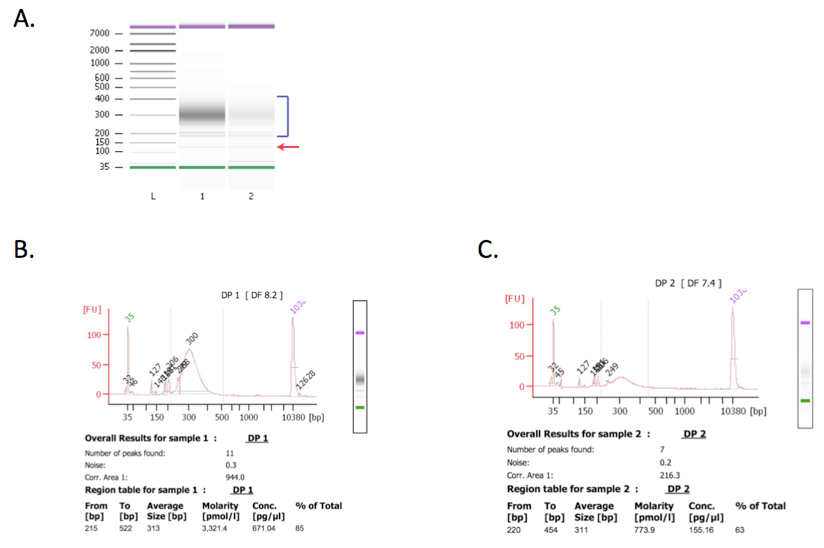
Figure 2. Bioanalyzer results. Libraries with higher concentration perform better in Illumina sequencing. Bioanalyzer analysis of libraries before sequencing is an important quality control step. Both libraries were in the expected size range (as indicated by blue bracket in panel A) and therefore suitable for sequencing. Library 1 (with higher molarity) was subjected to an additional clean up step using AMPure XP beads in order to reduce the amount of residual adapters (indicated by a red arrow in panel A, and panel B). Library 2 (panel C) was not cleaned up because it was at a low concentration and additional clean up might have caused loss of enough library such that sequencing would not be possible. Sequencing resulted in 12,489,378 and 3,824,500 total non-redundant reads for library 1 and 2, respectively.
Recipes
- 50x TAE (1 L)
242 g Tris base
57.1 ml glacial acetic acid
100 ml 0.5M EDTA (pH 8)
Acknowledgments
This work was supported by the NSF (MCB 1121952) and an award to MG from The Pew Charitable Trust’s Pew Scholars Program in the Biomedical Sciences.
This protocol was adapted from Pignatta et al. (2014).
References
- Groszmann, M., Greaves, I. K., Albertyn, Z. I., Scofield, G. N., Peacock, W. J. and Dennis, E. S. (2011). Changes in 24-nt siRNA levels in Arabidopsis hybrids suggest an epigenetic contribution to hybrid vigor. Proc Natl Acad Sci U S A 108(6): 2617-2622.
- Krueger, F. and Andrews, S. R. (2011). Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 27(11): 1571-1572.
- Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., Marth, G., Abecasis, G. and Durbin, R. (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25(16): 2078-2079.
- Quinlan, A. R. and Hall, I. M. (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26(6): 841-842.
- Schultz, M. D., Schmitz, R. J. and Ecker, J. R. (2012). 'Leveling' the playing field for analyses of single-base resolution DNA methylomes. Trends Genet 28(12): 583-585.
- Pignatta, D., Erdmann, R. M., Scheer, E., Picard, C. L., Bell, G. W. and Gehring, M. (2014). Natural epigenetic polymorphisms lead to intraspecific variation in Arabidopsis gene imprinting. Elife 3: e03198.
Article Information
Copyright
![]() Pignatta et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
Pignatta et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Pignatta, D., Bell, G. W. and Gehring, M. (2015). Whole Genome Bisulfite Sequencing and DNA Methylation Analysis from Plant Tissue. Bio-protocol 5(4): e1407. DOI: 10.21769/BioProtoc.1407.
- Pignatta, D., Erdmann, R. M., Scheer, E., Picard, C. L., Bell, G. W. and Gehring, M. (2014). Natural epigenetic polymorphisms lead to intraspecific variation in Arabidopsis gene imprinting. Elife 3: e03198.
Category
Systems Biology > Epigenomics > DNA methylation
Plant Science > Plant molecular biology > DNA > DNA sequencing
Molecular Biology > DNA > DNA modification
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.