- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Infectious Focus Assays and Multiplicity of Infection (MOI) Calculations for Alpha-herpesviruses

Published: Vol 4, Iss 22, Nov 20, 2014 DOI: 10.21769/BioProtoc.1295 Views: 44528
Reviewed by: Valeria LullaAnonymous reviewer(s)
Original research article
The authors used this protocol in:

May 2014
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
Titration of viral stocks is a critical process before any experimental use of the virus. Here we describe an infectious focus assay for several alphaherpesviruses, a titration method for fluorescently labeled viruses, based on the original plaque assay. In addition, the calculation of multiplicity of infection (MOI) is presented.
Keywords: Multiplicity of infectionMaterials and Reagents
- Standard cell line [e.g. MeWo (ATCC® HTB-65 TM); ARPE19 (ATCC® CRL-2302TM); Vero (ATCC® CCL-81 TM)]
- Fluorescently labeled virus stock (genetically engineered VZV-66GFP, HSV1-gC-GFP and PRV-IE180-GFP were kindly provided by Prof. Paul R. Kinchington, University of Pittsburgh, Pittsburgh PA)
- Growth medium for the cells listed above (e.g. item #6, Fibroblast medium)
Note: However, it can be any other medium for other pairs of virus and cells. - 4% paraformaldehyde (EMS 15710) in phosphate buffered saline with divalent cations (e.g. Biological Industries, catalog number: 020201A )
- High glucose DMEM (Biological Industries, catalog number: 010551A )
- Fetal calf serum (Biological Industries, catalog number: 041271A )
- Glutamine (Biological Industries, catalog number: 030201 )
- Penicillin-streptomycin (Biological Industries, catalog number: 030311 )
- Agarose low EEO (Hispanagar, catalog number: D1500 )
- Fibroblast medium (see Recipes)
- Overlaying media for free virus (see Recipes)
- Crystal violet (Sigma-Aldrich, catalog number: C0775 ) (see Recipes)
Equipment
- 24-well tissue culture plates
- Incubator 37 °C, 5% CO2
- Epifluorescent inverted microscope
Procedure
- Cell-associated varicella zoster virus (VZV)
- Grow MeWo or ARPE19 cells in a 24 well plate until they reach 90% confluence (~300,000 or 200,000 cells/well when seeding ~24 h before the infection, respectively). Use 500 µl/well volume to grow and infect the cells. The use of MeWo cells for VZV is documented in Heineman and Cohen (1994), Eisfeld et al. (2007) and Markus et al. (2011).
- Prepare 10-fold serial dilutions of VZV-infected MeWo cells. Typically, the range of the dilutions is 10-1 to 10-6 of the initial stock. The volume inoculated to each well can range up to 500 µl.
- Add the infected cells to the 24-well plate containing the 90% confluent naïve cell cultures in triplicate and incubate at 37 °C for at least 2 h. There is no special need to replace the medium after the addition of the inoculum, therefore the infected-cells can be left overnight as well. No agar overlay is required as no secondary plaques are formed by VZV, which does not release virions into the overlaying medium.
- Using a 10x objective with a fluorescent microscope, count all the fluorescent foci (groups of fluorescent cells) in each well after 2-5 days. Take care not to count the same fluorescent focus twice, and screen all the wells in the same order. This process can be automated with a microscope equipped with a computerized stage and image analysis software. If all fluorescent foci have developed into plaques then units used for the final virus concentration are plaque forming units (PFU).
- To calculate the PFU/ml, use the average number of foci in the wells with an input virus dilution resulting in 10-100 foci. For example, if the 10-3 dilution resulted in 24, 26 and 30 fluorescent foci per well, and the inoculated amount was 100 µl, then the stock concentration is

- Grow MeWo or ARPE19 cells in a 24 well plate until they reach 90% confluence (~300,000 or 200,000 cells/well when seeding ~24 h before the infection, respectively). Use 500 µl/well volume to grow and infect the cells. The use of MeWo cells for VZV is documented in Heineman and Cohen (1994), Eisfeld et al. (2007) and Markus et al. (2011).
- Cell-free VZV
- Grow ARPE19 cells in a 24 well plate until they reach 90% confluence (~200,000 cells/well when seeding ~24 h before the infection). These cells are more easily infected at low VZV concentrations as compared to MeWo or MRC5 cells (Schmidt-Chanasit et al., 2008; Pasdeloup et al., 2013). Use 500 µl/well volume to grow and infect the cells.
- Prepare 5-fold serial dilutions of cell-free VZV in high glucose DMEM. In our experience, diluting the virus in growth medium for cells which are not sensitive to the presence of serum does not seem to affect viral infectivity. Typically, the range of the dilutions is 1:5 to 1:625 of the initial stock.
- Inoculate the ARPE19 cells with 100-150 µl/well of the viral dilutions in triplicates.
- In a CO2 incubator, let the virus adsorb to the cell monolayer for 2 h, agitating occasionally.
- Remove the inoculum and add 0.5 ml pre-warmed growth media per well. No agar overlay is required since no secondary plaques are formed by VZV, which does not release virions into the overlaying medium.
- Monitor the cells for fluorescent foci formation. Usually, 5 days are sufficient for the foci to be easily countable, but this can vary between viruses and experiments.
- Using a 10x objective with a fluorescent microscope, count all the fluorescent foci (groups of fluorescent cells) in each well after 3-5 days. Take care not to count the same fluorescent focus twice, and screen all the wells in the same order. This process can be automated with a microscope equipped with a computerized stage and image analysis software. If all fluorescent foci have developed into plaques then units used for the final virus concentration are PFU.
- To calculate the PFU/ml, use the average number of foci in the wells with an input virus dilution resulting in 10-100 foci. For example, if the 1:25 dilution resulted in 25, 26 and 30 fluorescent foci, and the inoculate was 100 µl, then the stock concentration is

- Grow ARPE19 cells in a 24 well plate until they reach 90% confluence (~200,000 cells/well when seeding ~24 h before the infection). These cells are more easily infected at low VZV concentrations as compared to MeWo or MRC5 cells (Schmidt-Chanasit et al., 2008; Pasdeloup et al., 2013). Use 500 µl/well volume to grow and infect the cells.
- Cell-free PRV and HSV1
- Grow Vero cells (green African monkey kidney cells) in a 24 well plate until they reach 90% confluence (~400,000 cells/well when seeding ~24 h before the infection). The use of Vero cells for HSV1 and PRV is documented in Moffat et al. (1998), Nicola and Straus (2004) and Pasdeloup et al. (2013).
- Prepare 10-fold serial dilutions of the virus in high glucose DMEM. Typically, the range of the dilutions is 10-2 to 10-7 of the initial stock. Keep the dilutions on ice.
- Inoculate the Vero cells with 100 µl/well of the virus in quadruplicates.
- Let the virus adsorb to the cell monolayer for 1 h in a CO2 incubator, agitating every 15 min.
- Remove the inoculum and cover the cells with 1 ml of overlaying medium containing 0.3% agarose per well. Work quickly so that the media will not start to solidify before its addition to the cells.
- Monitor the cultures for formation of fluorescent foci, usually less than 4 days.
- Using a 10x objective with a fluorescent microscope, count all the fluorescent foci (groups of fluorescent cells) in each well after 3-5 days. Take care not to count the same fluorescent focus twice, and screen all the wells in the same order. This process can be automated with a microscope equipped with a computerized stage and image analysis software. If all fluorescent foci have developed into plaques then units used for the final virus concentration are PFU.
- To calculate the PFU/ml, use the average number of foci in the wells with an input virus dilution resulting in 10-100 foci. For example, if the 10-4 dilution resulted in 40, 43 and 45 fluorescent foci per well, and the inoculated amount was 100 µl, then the stock concentration is

- If virus without fluorescent label are used, or it is desirable to count plaques rather than fluorescent loci, plaques are visualized by staining cultures with 0.1% crystal violet: Carefully lift the overlaying agar medium using a spatula, taking care to not disturb the cell monolayer. Fix the cells with 100 µl 4% PFA in PBS for 30 min at room temperature. Wash twice with PBS, and add 0.1% crystal violet solution for 10 min. Carefully wash with water until the liquid no longer has traces of blue (usually about 5 rinses).
- Count the plaques and calculate the PFU/ml as above.
- Grow Vero cells (green African monkey kidney cells) in a 24 well plate until they reach 90% confluence (~400,000 cells/well when seeding ~24 h before the infection). The use of Vero cells for HSV1 and PRV is documented in Moffat et al. (1998), Nicola and Straus (2004) and Pasdeloup et al. (2013).
- Multiplicity of infection (MOI) calculation
- In general, the equation of the multiplicity of infection is the following:

- In order to calculate the MOI, you should first determine the number of cells you are infecting, and the titer of the virus inoculated on them.
- For example, if 2.5 x 104 human foreskin fibroblasts are seeded in each well of a 24 well-plate, and 250 µl of 104 PFU/ml VZV were used to infect it, the MOI is

- In general, the equation of the multiplicity of infection is the following:
Representative data
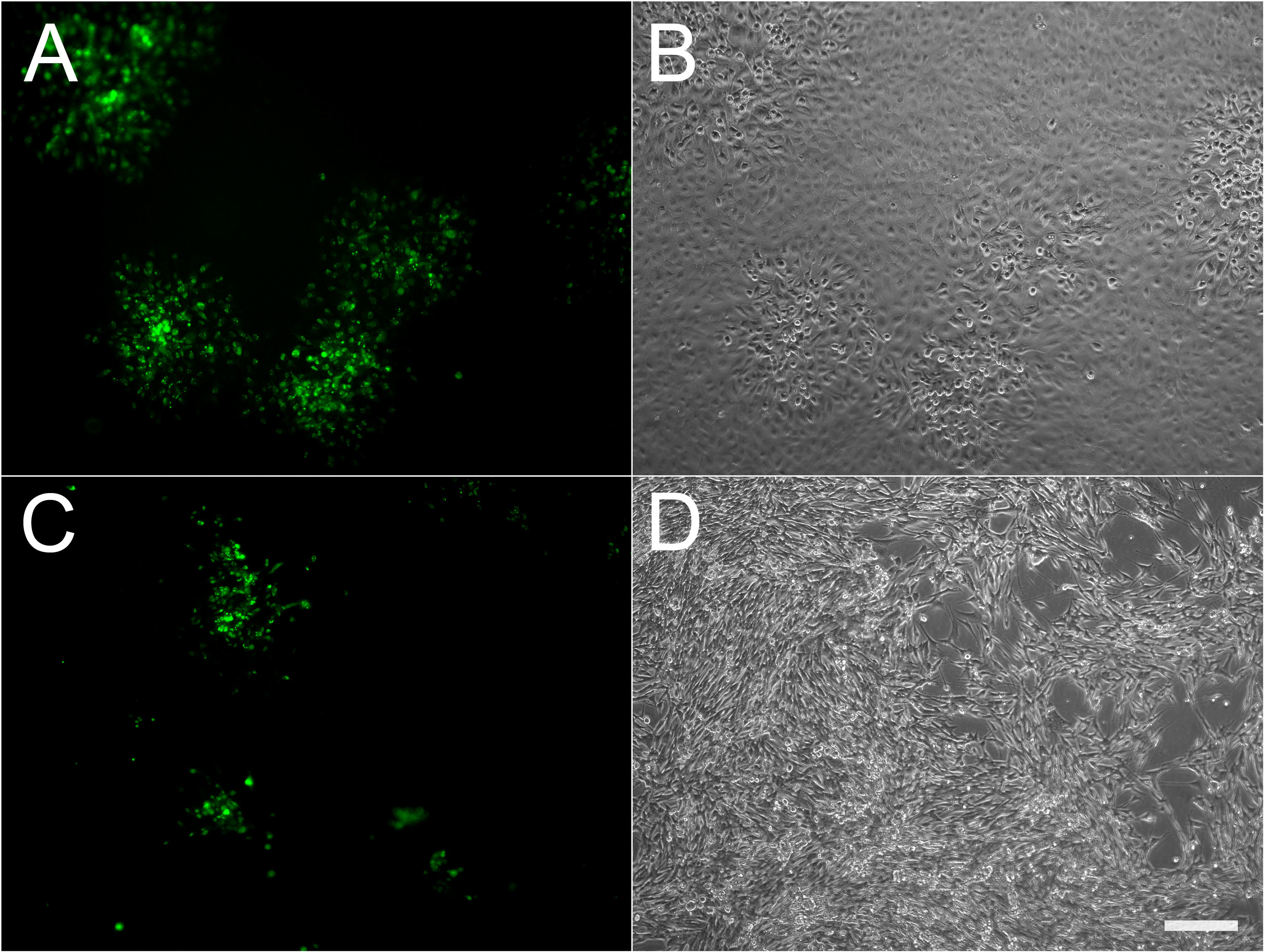
Figure 1. Microscopic appearance of infectious foci using fluorescent virus. ARPE19 (A, B) or MeWo (C, D) cells were infected with cell-associated VZV-23GFP. Both fluorescence (A, C) and phase contrast (B, D) images of the same fields are shown. Scale bar =200 µm
Figure 2. Plaque assay for HSV1 gCGFP using crystal violet staining. 4 days after infection, cells were fixed and stained as described in the text. Viral concentration is decreasing from left to right. Crystal-violet stains the cells, which allows the visualization of the clear plaques. An arrow points to the dilution which yielded quantifiable plaques. Control, uninfected wells did not yield any detectable plaques (uninfected).
Recipes
- Fibroblast medium
For 500 ml, combine 450 ml of high glucose DMEM with 50 ml fetal bovine serum. Supplement with 2.5 ml glutamine and 5 ml penicillin/streptomycin - Overlaying media for free virus
In advance, prepare a stock of 1.2% pure agarose for electrophoresis in PBS by autoclaving. Pre-heat the required amount of medium to 37 °C.
Right before use, re-boil the agar in a microwave oven to liquefy it. Dilute the agar with the growth medium at a ratio of 1 agar volume to 3 medium volumes, e.g., add 6 ml agar to 18 ml medium in a 50 ml tube. This will result in 0.3% agar in medium which is sufficient to cover one 24-well plate.
Keep the overlaying media tube inside a beaker with ~40 °C water, to prevent solidification. - Crystal violet
Dissolve 0.1% crystal violet in 10% ethanol (e.g., 5 ml ethanol and 45 ml H2O)
First add the powder to the appropriate amount of 100% ethanol to completely dissolve it, and then add the required amount of water to generate 10% ethanol.
Acknowledgments
The HSV1 titration was based on an unpublished protocol of Dr. Maya Ilouze (Sheba Medical Center, Tel-HaShomer, Israel). We gratefully acknowledge the support of grant from the Israel Science Foundation 238/11 and a donation from the Maximillian Goode Foundation.
References
- Eisfeld, A. J., Yee, M. B., Erazo, A., Abendroth, A. and Kinchington, P. R. (2007). Downregulation of class I major histocompatibility complex surface expression by varicella-zoster virus involves open reading frame 66 protein kinase-dependent and -independent mechanisms. J Virol 81(17): 9034-9049.
- Heineman, T. C. and Cohen, J. I. (1994). Deletion of the varicella-zoster virus large subunit of ribonucleotide reductase impairs growth of virus in vitro. J Virol 68(5): 3317-3323.
- Markus, A., Grigoryan, S., Sloutskin, A., Yee, M. B., Zhu, H., Yang, I. H., Thakor, N. V., Sarid, R., Kinchington, P. R. and Goldstein, R. S. (2011). Varicella-zoster virus (VZV) infection of neurons derived from human embryonic stem cells: direct demonstration of axonal infection, transport of VZV, and productive neuronal infection. J Virol 85(13): 6220-6233.
- Moffat, J. F., Zerboni, L., Kinchington, P. R., Grose, C., Kaneshima, H. and Arvin, A. M. (1998). Attenuation of the vaccine Oka strain of varicella-zoster virus and role of glycoprotein C in alphaherpesvirus virulence demonstrated in the SCID-hu mouse. J Virol 72(2): 965-974.
- Nicola, A. V. and Straus, S. E. (2004). Cellular and viral requirements for rapid endocytic entry of herpes simplex virus. J Virol 78(14): 7508-7517.
- Pasdeloup, D., Labetoulle, M. and Rixon, F. J. (2013). Differing effects of herpes simplex virus 1 and pseudorabies virus infections on centrosomal function. J Virol 87(12): 7102-7112.
- Schmidt-Chanasit, J., Bleymehl, K., Rabenau, H. F., Ulrich, R. G., Cinatl, J., Jr. and Doerr, H. W. (2008). In vitro replication of varicella-zoster virus in human retinal pigment epithelial cells. J Clin Microbiol 46(6): 2122-2124.
- Sloutskin, A. and Goldstein, R. S. (2014). Laboratory preparation of Varicella-Zoster Virus: concentration of virus-containing supernatant, use of a debris fraction and magnetofection for consistent cell-free VZV infections. J Virol Methods 206: 128-132.
Article Information
Copyright
© 2014 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Sloutskin, A. and Goldstein, R. S. (2014). Infectious Focus Assays and Multiplicity of Infection (MOI) Calculations for Alpha-herpesviruses. Bio-protocol 4(22): e1295. DOI: 10.21769/BioProtoc.1295.
Category
Microbiology > Microbial cell biology > Cell imaging
Microbiology > Microbe-host interactions > Virus
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.