- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Phagolysosomal Trafficking Assay

Published: Vol 4, Iss 13, Jul 5, 2014 DOI: 10.21769/BioProtoc.1163 Views: 17167
Reviewed by: Hong-guang XiaKanika GeraAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Oct 2013
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
Phagolysosomal trafficking is an important innate defense pathway that clears microbes by delivering them to lysosomes, the degradative compartment of the cell. Mycobacterium tuberculosis (Mtb), the causative agent of tuberculosis, subverts this host defense mechanism by arresting maturation of the phagosome. The ability of Mtb to arrest its delivery to the lysosome can be demonstrated by the prolonged co-localization of bacteria containing phagosomes/vacuole with early phagosomal markers [such as, Ras- related proteins in the brain 5 (Rab5) and Transferrin receptor (TfR)], and a failure to acquire late phagosomal and lysosomal markers (such as Rab7 and LAMP1) (Deretic and Fratti, 1999, Mehra et al., 2013). Here, a protocol is outlined for infection of macrophages with mycobacterial species like pathogenic Mtb, vaccine strain Mycobacterium bovis- bacillus Calmatte- Guérin (BCG) and rapidly dividing non-pathogenic Mycobacterium smegmatis (Msmeg), followed by indirect-immunofluorescence microscopy to visualize host vacuolar markers. Thereafter, automated quantification of degree of co-localization between mycobacteria and host vacuolar markers like TfR and LAMP1 is done by processing the binary images of bacteria using mathematical tools. This results in quantification of the mean fluorescence intensity (MFI) of these host markers directly around the bacteria/bacterial clusters with increased sensitivity relative to when done manually. By manipulating host or pathogen, this assay can be used to evaluate host or bacterial determinants of intracellular trafficking. The basic method can be applied to studying trafficking of other bacteria or particles like beads, although the kinetics of infection and phagosome maturation will depend upon the phagocytic cargo. The mathematical analysis tools are available in many standard imaging analysis programs. However, any adaption for similar analysis should be confirmed by the individual user with their imaging and analysis platform.
Keywords: Macrophage infectionsMaterials and Reagents
Note: All work with live Mtb must be performed in a Biosafety Level 3 (BSL3) facility according to institutional standards of practice.
- Macrophages, either primary macrophages, such as C57BL/6 bone marrow-derived macrophages (BMDMs) or a macrophage cell line (such as RAW264.7)
Note: BMDMs can be isolated as described (Banaiee et al. 2006; Nagabhushanam et el., 2013). RAW264.7 cells can be purchased from ATCC (ATCC, catalog number: TIB-71 ). - L929 cells (ATCC, catalog number: CCL-1 )
- Dulbecco’s Modified Eagle Medium (DMEM) (Life Technologies, Gibco®, catalog number: 11965 )
- Fetal Bovine Serum (FBS) (heat inactivated) (Life Technologies, Gibco®, catalog number: 10082147 )
- 1 M HEPES solution (Life Technologies, Gibco®, catalog number: 15630-056 )
- 200 mM L-glutamine (Life Technologies, Gibco®, catalog number: 25030-081 )
- Penicillin-Streptomycin solution (10,000 U/ml) (Life Technologies, Gibco®, catalog number: 15140-122 )
- Phosphate buffered saline (PBS) (Life Technologies, Gibco®, catalog number: 10010-023 )
- Eight well Permanox chamber slide (Thermo Fisher Scientific, Nunc Lab-Tek Chamber Slides, catalog number: 177445 )
- Eight well chamber coverglass (Thermo Fisher Scientific, Nunc Lab-Tek Chamber coverglass, catalog number: 155411 )
- Paraformaldehyde (PFA) (Sigma-Aldrich, catalog number: P6148 )
- Bovine serum albumin (BSA) (fraction V) (Thermo Fisher Scientific, catalog number: BP1600 )
- Detergents: Saponin (Sigma-Aldrich, catalog number: 47036 ), Triton- X100 (Sigma-Aldrich, catalog number: X100) and/or Tween-20 (Thermo Fisher Scientific, catalog number: BP337 )
- Primary antibodies to detect host cellular markers
For example, recycling endosomes and early phagosomes can be labeled with mouse anti-transferrin receptor (anti-TfR) antibody (Life Technologies, InvitrogenTM, catalog number: 136800 ); Late endosomes and lysosomes stain with rabbit anti-LAMP1 antibody (Abcam, catalog number: 24170 ).
Notes:- If Mtb infected slides are to be removed from the BSL3 for imaging, the antibodies chosen need to work after fixation cum sterilization methods like long fixation as mentioned below in step A11, note a. Some of the commercially available antibodies may loose recognition or weakly recognize their epitopes after long fixation.
- It is critical that polyclonal antibodies were not raised in animals given Freund’s adjuvant, as then they will directly recognize Mtb in addition to whatever cellular marker they were raised against. All antibodies should be tested to verify that they do not directly recognize Mtb.
- If Mtb infected slides are to be removed from the BSL3 for imaging, the antibodies chosen need to work after fixation cum sterilization methods like long fixation as mentioned below in step A11, note a. Some of the commercially available antibodies may loose recognition or weakly recognize their epitopes after long fixation.
- Secondary antibodies for immunofluorescence
Secondary antibodies are available against different species and in different colors and user may choose depending on the primary antibodies being used. They are adsorbed against multiple species to minimize species cross reactivity during immunostaining. For example, Goat anti- mouse Alexa 594 (Life Technologies, Molecular Probes®, catalog number: A11032) and Goat anti-rabbit Alexa 594 (Life Technologies, Molecular Probes®, catalog number: A11037 ).
Of note, Mtb exhibits autofluorescence, with an emission maximum at 475 nm when excited at 405 nm, and thus are visualized by many DAPI filters (Patiño et al., 2008). Therefore, secondary antibodies should be chosen that do not fluoresce in this range. - Lysotracker Red DND-99 (1 mM stock in DMSO) (Life Technologies, Molecular Probes®, catalog number: L-7528 )
Note: Lysotracker dyes are available in different colors and one may choose depending on the color requirement. - Dextran (TexasRed, 10,000 MW, Lysine fixable) (Life Technologies, Molecular Probes®, catalog number: D-1863 ) (make 25 mg/ml stock in PBS, stored in dark in -20 °C)
Note: Dextran is available in different colors and molecular weights and again one may choose depending on requirement and desired goals of the experiment. - Vectashield mounting media (Vector Laboratories, catalog number: H-1000 )
- Middle brook 7H9 broth (Difco, catalog number: 271310 )
- Albumin-dextrose-catalase (ADC) (BD, catalog number: 212352 )
- Oleic-albumin-dextrose-catalase (OADC) (BD, catalog number: 212351 )
- Nail polish (clear)
- Immersion oil (Microscope 50CC Immersion oil) (e.g. Nikon Corporation, catalog number: IB-MA-MXA20234 )
- 4% paraformaldehyde solution in PBS (see Recipes)
- DMEM complete media (see Recipes)
- DMEM/L929 complete media (see Recipes)
- L-Cell conditioned media (see Recipes)
- 2% BSA in PBS containing 0.1% saponin (see Recipes)
- 2% BSA in PBS containing 0.1% triton X-100 (see Recipes)
- 7H9 complete media (see Recipes)
- Fixative (see Recipes)
- Blocking solution (see Recipes)
Equipment
- Spectrophotometer (measure the OD600 i.e. optical density at wavelength of 600 nm of the mycobacterial cultures using cuvettes)
- Disposable 1.5 ml cuvettes (Perfector Scientific, catalog number: 9003 )
- Disposable sterile filter system (500 ml, 0.22 µm pore size) (Corning, catalog number: 430758 )
- 30 ml square media bottles (Thermo Fisher Scientific, Nalgene, catalog number: NE/2019-0030 )
- 50 ml, 15 ml falcon tubes (with plug seal caps) (Corning, catalog numbers: 430052 and 430290 )
- Coverslip (22 x 50 mm, thickness#1, 0.13-0.17 mm) (Thermo Fisher Scientific, catalog number: 12-545C )
- Centrifuge with swinging bucket rotor for spinning down bacterial cultures (for example, Beckman Coulter, model: Allegra X-15R ; bench top centrifuge with SX4750 rotor)
Note: Mtb cultures should be handled in BSL3 facility according to institutional standards of practice. - 37 °C shaker incubator with aerosol containment units for Mtb liquid cultures
- Beckman aerosolve canisters for centrifuging mycobacterial cultures in falcon tubes (e.g. Beckman Coulter, catalog number: BK359232 )
- Multiwell-Plate Carrier Covers (e.g. Beckman Coulter, more on this link https://www.beckmancoulter.com/wsrportal/techdocs?docname=GX-TB-012)
- 37 °C shaker incubator with aerosol containment units for Mtb liquid cultures
- Epifluorescence microscope [e.g. Nikon Eclipse TiE/B model equipped with 60x; Plan-Apochromat, NA 1.4 oil immersion objective, Ti Z drive, high resolution monochrome charge-coupled device (CCD) digital camera; Photometric Cool SNAP HQ2 and appropriate filter sets for DAPI, FITC and TexasRed channel]
Software
- Nikon Imaging Software-Elements Advanced Research (NIS-Elements AR) version 3.2 software with deconvolution module
- Graph Pad Prism software
Procedure
- Infection of macrophages
Notes:- All work with live Mtb to setup infection (steps A1 and A3-11 below) should be done in bio-safety level 3 (BSL3) facility.
- BCG and Msmeg cultures should be handled outside BSL3 facility but in biosafety class II cabinets or as per institutional standards of practice.
- Centrifugation of Mtb and BCG cultures (steps A4-6 below) and infected samples (step A9 below) should be done using suitable aerosol canisters and multiwell plate cover or as per institutional standards of practice. These canisters and multiwell plate carriers with covers should be opened for loading and unloading of Mtb/BCG culture containing falcons or infected plates in the bio-safety class II cabinets during centrifugation.
- Starting with frozen bacterial stocks (prepared from mid-log phase culture i.e. culture with OD600 between 0.5 to 1.0 units frozen in 18% glycerol at -80 °C) inoculate liquid cultures of mycobacteria in 10 ml of 7H9 complete media in 30 ml square media bottles. Include antibiotics as appropriate (for example, to select plasmids containing GFP with kanamycin as the selection marker, include kanamycin at 5 µg/ml). Incubate at 37 °C with shaking at 90-110 rpm in aerosol containment units. Mtb and BCG double approximately every 20 h in 7H9 complete media at 37 °C so the cultures will take few (~4-5) days to reach mid-log phase. The cultures maybe diluted if required into fresh media (e.g. if antibiotic used is prone to degradation during culture) at appropriate intervals. Msmeg doubles approximately every 3 h and so dilute an overnight grown culture in the morning to reach mid-log phase during the day to use for infection. The timing will depend upon the particular strain, conditions, and starting inoculum.
- Plating of macrophages should be done one day prior to infection in bio-safety class II cabinet in the tissue culture lab. Plate the cells in 250 µl of either DMEM or DMEM/L929 complete media per well in 8 well chamber slide. BMDMs can be plated at a density of 1 x 105 per well. If RAW264.6 macrophage cell line is used, it can be seeded with a density of 6 x 104 per well in DMEM complete media. Incubate the cells in 37 °C incubator with 5% CO2 atmosphere.
On the day of Mtb infection, transfer the slides to the 37 °C incubator with 5% CO2 atmosphere in the BSL3 facility before proceeding further.
Of note, coverslips placed in 24 well plate can also be used for plating the macrophages for infection. However, 8 well chamber slide and cover glass offer sterile and RNase free plating conditions with minimal use of reagents. In addition, multiple conditions can be tested with minimal well to well variation like different host markers can be knocked down prior to infection to see the role of host protein in phagolysosomal trafficking or multiple bacterial strains can be infected in different wells of the same slide. - Measure the OD600 of 1ml of the culture in cuvette using spectrophotometer. Dilute the culture if required so as to have the OD600 of the culture between 0.5 to 1.0 OD600 on the day of infection of macrophages.
- Transfer culture into 15 ml falcon tube and spin at 1,500 x g for 5 min at room temperature.
- Remove the supernatant and resuspend the pellet in 5 ml of PBS and again spin at 1,500 x g for 5 min at room temperature. Repeat it once more to remove carried over Tween 80 from 7H9 complete media.
- Resuspend the pellet in 5 ml of DMEM complete media for RAW 264.7 macrophages and DMEM/L929 complete media for BMDM and transfer it to 50 ml falcon. Spin at low speed of 450 x g for 5 min at room temperature to remove bacterial clumps by pelleting. Transfer the supernatant carefully avoiding the pellet to a fresh 15 ml falcon tube. The supernatant is devoid of mycobacterial clumps with substantial single cell population and is therefore used to infect macrophages.
Note: It is not unusual to remove ~ 90% of the mycobacteria by pelleting in this step. For example, if at step A3, the OD600 of a 10 ml culture of Mtb is 0.5 then expect the supernatant after low speed spin to have an OD600 ~ 0.05 to 0.1 depending on the degree of clumping. - Determine the OD600 of the supernatant harvested in step A6. Convert the OD600 to number of bacteria. This value will vary depending upon the shape, size and internal light absorbing components of the bacteria and may be distinct for different strains of the same species. One should predetermine the conversion factor. For example, a conversion factor such as OD600 of 1.0 = 500 x 106 colony forming units (CFU)/ml can be determined as below:
- a.A starting culture of “x” OD600 is serially diluted and a volume of each dilution is plated to give CFUs.
- The dilution which give colonies in countable range and the number of colonies for that dilution are used to calculate “y” CFU/ ml using the formula:
CFU/ml = (# colonies) * (dilution factor)/ (volume plated in ml). - This gives x OD600 = y CFU/ml. Now OD600 of 1 = y/x CFU/ml.
- a.A starting culture of “x” OD600 is serially diluted and a volume of each dilution is plated to give CFUs.
- Calculate the volume of the bacterial suspension prepared in step A6 that is required to infect macrophages at a multiplicity of infection (MOI) of ~ 1-10. Add this volume to each the well. If necessary, make a dilution so that at least 10-50 µl is added to each well in a total volume of 200 µl. Several MOIs should be tested and the optimal MOI will depend upon details of the experimental conditions.
- Spin the slide at 50 x g for 2 min at room temperature to synchronize the infection using multiwell plate carriers with covers in the swing bucket rotor. Incubate in 37 °C incubator with 5% CO2 atmosphere for 3 h. Different strains may differ in infectivity of the macrophages.
- After 3 h, remove uninternalized bacteria by washing three times with 300 µl pre-warmed PBS. Add 300 µl of DMEM or DMEM/L929 (for RAW cells or BMDMs, respectively) and incubate in 37 °C incubator with 5% CO2 atmosphere. It is possible to perform this step after a shorter time period, although bacterial uptake will be lower.
- At desired time points [such as 3 h post-infection (hpi), 12 hpi, and 24 hpi], remove the media. Fix in 1% PFA/PBS at 4 °C overnight for Mtb infected samples or as per institutes bio-safety guidelines before removing them from the BSL3. Fix in 4% PFA/PBS at room temperature, 15 min, for Msmeg and BCG infected samples.
Notes:- As per biosafety rules, Mtb infected slides should be sterilized before taking them out of BSL3 for immunostaining and imaging (Schwebach et al., 2001). This can be achieved by long fixation method; fixing in 1% PFA/PBS, overnight (minimum of 12 h) at 4 °C or as per institutional’s standards of practice.
- For alternate fixatives, like methanol or acetone, the dishes for plating macrophages and infection should be compatible with organic solvents.
- As per biosafety rules, Mtb infected slides should be sterilized before taking them out of BSL3 for immunostaining and imaging (Schwebach et al., 2001). This can be achieved by long fixation method; fixing in 1% PFA/PBS, overnight (minimum of 12 h) at 4 °C or as per institutional’s standards of practice.
- All work with live Mtb to setup infection (steps A1 and A3-11 below) should be done in bio-safety level 3 (BSL3) facility.
- Staining for immunofluorescence microscopy
For immunofluorescence (IF) microscopy, one has to pre-optimize the conditions for immunostaining which includes fixatives (see step A11, notes a-b), detergent to permeablize the cells (triton X-100, saponin or tween-20), blocking and dilution for each primary antibody before performing co-localization experiments (Goldenthal et al., 1985). During standardization, the specificity of immunostaining can be confirmed by silencing the host marker by RNA interference (RNAi). It is important to include controls that would help in setting acquisition exposures and also serve as a control for non-specific staining during fluorescence microscopy as explained further in step C21. For Mtb infected samples, primary antibodies for a specific host marker should be screened for recognition of specific epitopes for optimal sensitivity in macrophages which have been fixed for long to sterilize Mtb which is in contrast to BCG and Msmeg infected samples which are fixed for short (as described in step A11 of the protocol).- Wash out the fixative well with 300 µl of PBS three times.
- Blocking: Incubate in blocking buffer for 1 h at room temperature. For example, blocking buffer can be 2% BSA in PBS containing 0.1% detergent optimized for the primary antibody. The detergent permeablizes the plasma membrane so that the antibody can enter the cell, and BSA blocks to prevent non-specific staining.
For the commercially available primary antibodies mentioned in materials and reagents, transferrin receptor (TfR) and LAMP1, add 200 µl of 2% BSA in PBS-0.1% triton X-100 and 2% BSA in PBS-0.1% saponin, respectively, to the corresponding wells to block. - Primary antibody: Add 150 µl of the diluted primary antibody to the well and incubate overnight at 4 °C. Dilute the primary antibody in the 2% BSA in PBS-0.1% detergent as standardized. For the primary antibodies mentioned in materials and methods, dilute TfR (1: 250 v/v) and LAMP1 (1: 1,000 v/v) in 2% BSA in PBS-0.1% triton X-100 and 2% BSA in PBS-0.1% saponin, respectively.
- Wash with PBS-0.1% detergent three times, each time for 5 min at room temperature to remove excess and non-specifically bound antibody e.g. use PBS -0.1 % triton X-100 for transferrin receptor and PBS -0.1 % saponin for LAMP1 antibodies used in step B14.
- Secondary antibody: Depending on species in which primary antibody was raised, use the appropriate anti-species (mouse or rabbit) secondary antibody labeled with appropriate fluorophore. Add 150 µl of the diluted secondary antibody and incubate for 1 h at room temperature in the dark to prevent bleaching of the fluorophore. Dilute the secondary antibody 1: 250 in 2% BSA in PBS-0.1% detergent e.g. use anti-mouse Alexa 594 in 2% BSA in PBS-0.1% triton X-100 to detect anti-TfR and dilute anti-rabbit Alexa 594 in 2% BSA in PBS-0.1% saponin to detect anti-LAMP1.
- Repeat step B15. Protect from light to prevent photo-bleaching of the secondary antibody.
- Remove the chambers from the top of the slide by gentle upward pressure and peel off the rubber gasket (see Figure 1).
- Put ~2 µl of anti-fade in the center of each of the eight well on the slide. Mount a clean rectangular coverslip and seal the sides of the coverslip with nail polish.
- Although not described in detail here, alternatively, it is also possible to do live cell staining of lysosomes using lysotracker dyes or loading them with fluorescent dextran (e.g. TexasRed-dextran). The kinetics of live staining has mainly two variables, concentration and time and depending on the desired goals one may best design the experiment to couple it with infection. A brief understanding towards it usage is provided:
- Lysotracker is a cell permeable, acidotrophic stain that can be used to label lysosomes in macrophages without the need for fixation. Staining conditions vary and users may standardize concentration of the dye and time of staining (30 min to 2 h) depending on the actual experiment.
- Plate the macrophages and infect them as described in section A. At the last 30 min of the desired time point post-infection of macrophages, remove media from well, add 150 µl of 200 nM lysotracker per well and incubate at 37 °C. Protect the samples from light hereafter to prevent photobleaching. Turn off the light in the biosafety cabinet while working with the stained sample to prevent photo-bleaching.
- Wash the well three times with PBS to remove excess of the lysotracker.
- Add PBS and quickly proceed for image acquisition with the fluorescence microscope. Further incubation in dye free media often leads to fading of the signal and cell blebbing.
Note: Msmeg and BCG infected macrophage samples can be directly taken for live image acquisition and so macrophages in this case should be plated on 8 well chamber cover glass instead of the slide so that the chambers do not have to be removed. In case of Mtb infected macrophages, after live labeling with lysotracker, the samples should be fixed as per institutes bio-safety guidelines before taking them out of BSL3 for imaging. They can then be washed with PBS to remove fixative and mounted with the anti-fade.
- Plate the macrophages and infect them as described in section A. At the last 30 min of the desired time point post-infection of macrophages, remove media from well, add 150 µl of 200 nM lysotracker per well and incubate at 37 °C. Protect the samples from light hereafter to prevent photobleaching. Turn off the light in the biosafety cabinet while working with the stained sample to prevent photo-bleaching.
- TexasRed-dextran (10 kD) is freely permeable to the endocytic and lysosomal vesicular network of the cell and so can be used to label lysosomes before infection by pre-loading the macrophages:
- Plate the macrophages as in step A2. Remove the plating media from the well and treat with 150 µl of 1 mg/ml solution of dextran (diluted in PBS) for 1h at 37 °C for cellular uptake by fluid phase endocytosis.
- Wash with pre-warm complete DMEM or DMEM/L929 complete media (as required) twice, observe under the microscope and if required wash more with pre-warmed PBS twice.
- Add back warm complete DMEM or DMEM/L929 complete media (as required) and incubate for 4 h in 37 °C incubator with 5% CO2 to chase the TexasRed-dextran to lysosomes. This chase period can be done overnight followed by infection the next day.
- Infect the macrophages and fix the samples at the desired time points post-infection.
- Plate the macrophages as in step A2. Remove the plating media from the well and treat with 150 µl of 1 mg/ml solution of dextran (diluted in PBS) for 1h at 37 °C for cellular uptake by fluid phase endocytosis.
- Lysotracker is a cell permeable, acidotrophic stain that can be used to label lysosomes in macrophages without the need for fixation. Staining conditions vary and users may standardize concentration of the dye and time of staining (30 min to 2 h) depending on the actual experiment.
- Wash out the fixative well with 300 µl of PBS three times.
- Image acquisition
Note: Appropriate training should be acquired before operating any fluorescence or confocal microscope for image acquisition as per individual or core facility guidelines.- For image acquisition (see Reference 8 for more details), it is important to have these controls:
- Fixed but unstained as a control for background autofluoresence of the cells and bacteria.
- Secondary alone control to check for non-specific staining.
- Single color controls in case of multi-color labeling experiment e.g. when doing dual immunolabeling of two different markers with compatible staining procedures in the same well. This is essential to collect images for each single color in all channels at exactly same settings as being used to acquire image of the multi-color sample. It helps to correct for the crosstalk (excitation for one dye with incident light indented for the other dye) and bleed-through (emission of one dye into detector for other dye).
- If single color controls indicate bleed through, it is best to do immunolabeling of only one marker in each well.
- Fixed but unstained as a control for background autofluoresence of the cells and bacteria.
- Images are acquired using the instructions specific to the fluorescence or confocal microscope at high magnification e.g. with 60x 1.4 NA oil immersion objective using Nikon Eclipse TiE/B microscope. Set optimal exposure levels for differential interference contrast (DIC) and required color channel (DAPI, GFP and/or TxRed) without saturating the pixel intensity. Using the auto exposure tool with single color controls is useful in estimating an exposure level to prevent cross-talk.
- After phagocytosis, mycobacteria with slender rod like morphology is mainly localized in vesicular structures/phagosomes in the cytoplasm of the cell up to 48 h (van der Wel et al., 2007). Mycobacteria is 0.2 to 0.5 µM in width with average width of 0.35 µM and so appropriate z-stacks should be obtained. A minimum of three fields with 10 to 15 macrophages per field should generally be acquired per well to generate a minimum of 100 regions of interest (ROI) per condition to use for calculation of statistical significance.
Note: When using the autofluorescence of Mtb in DAPI channel to visualize bacteria, it is advised to keep the exposure of the sample to exciting incident light at 405 nm to minimum since it quickly bleaches the autofluorescence of Mtb. - Deconvolute the images if required as in case of acquisition with Nikon Eclipse TiE/B epifluorescence microscope and correct for background on all images before analysis.
- For image acquisition (see Reference 8 for more details), it is important to have these controls:
- Image analysis
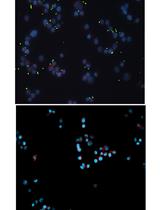
Endosomal/lysosomal markers localize around the phagosomal cargo (Fratti et al., 2001; Hava et al., 2008; Delaby et al., 2012; Mottola et al., 2014). In the illustrated example in Figure 2A, the host marker, transferrin receptor (TfR) is shown in grey and the phagosomal cargo, Mtb is in green. Here the degree of co-localization of the host markers and bacteria/bacterial clusters is variable between phagosomes (see phagosomes 1, 2, 3 and 4 in Figure 2A). Qualitative analysis done visually suggests phagosomes, 2 and 3 have high and phagosomes, 1 and 4 have low TfR staining around bacteria. For quantification of the host vacuolar staining around bacteria/bacterial clusters, I describe automated analysis done using NIS-Elements AR version 3.2 using Figure 2 as an example.- Create a binary image in the color channel of the bacteria (green). In binary images, the background will have 0 pixel intensity and the selected object (bacteria) will have a pixel intensity of 1. In the given example, Figure 2B, this can be achieved by “Thresholding” the intensity in the color channel of the bacteria. Additionally, in NIS Elements Advanced Research software, the “Autodetect all” tool can be used which works by detecting the intensity of the pixels under the cursor and so the cursor movement helps to mark all the bacteria in a given field.
- Using the binary image of the bacteria, “Dilate” three times to enlarge the marked bacteria in the active window. Copy this image with dilated bacteria to clipboard which is a reference binary image to be used later (Figure 2C).
- Now using the active window of the dilated bacterial image, “Erode” three times to shrink the enlarged area in step D26. This creates a current binary image to be used later (Figure 2D).
- Use the command “eXclusive OR” (XOR) function on the two images (Figure 2E), the reference and current binary from steps D26-27, respectively to generate a processed result binary image where “rings” are marked around bacteria/bacterial clusters (Figure 2F).
- In the active result binary image, converting the rings to regions of interest (ROIs) (see ROI IDs: 1-4 in Figure 2G).
- Open the image of the vacuolar marker (e.g. TfR) and measure the ROI in that channel to obtain the mean fluorescence intensity (MFI) for the ROIs (see Figure 2H).
Notes:- Validation of the analysis: The visual scoring of high co-localization on phagosomes 2 and 3 and less on phagosome 1 and 4 in Figure 2A correlates with automated quantification in Figure 2H with ROI ID: 2 having maximum mean intensity and ROI ID: 1 has the least mean intensity of fluorescence.
- The count number of three for dilation and erosion should be optimally determined for each phagosomal cargo by individual user on set of images to best capture the signal intensity of the vacuolar marker.
- Validation of the analysis: The visual scoring of high co-localization on phagosomes 2 and 3 and less on phagosome 1 and 4 in Figure 2A correlates with automated quantification in Figure 2H with ROI ID: 2 having maximum mean intensity and ROI ID: 1 has the least mean intensity of fluorescence.
- Plot the MFI for all the ROI on the Y-axis for each well. Graph display, statistical calculations and P values can be determined using Graph Pad Prism software.
Notes:- Morphology operators (dilate, erode) and Boolean logic operations are integrated as processing tools in many image analysis softwares for easy use. However, they are based on complex mathematical theories and so I refer to literature (Serra, 1987; Dougherty and Lotufo, 2003) to be careful with the interpretations of these processing parameters.
- Individual user should optimize automated analysis of co-localization of different host markers with phagosomal cargoes and tally the results with visual scoring to validate the analysis as described above in section D of the protocol.
- The results of automated analysis of a trafficking experiment (for example, co-localization of Mtb with TfR and LAMP1 at 3 and 24 hpi in Mtb infected macrophages) should be consistent with visual scoring done manually by a blinded observer.

Figure 1. Illustration showing detachment of the top chambers from the 8 well chamber slide
Remove the washing buffer from the wells (A) and insert blade as shown (B) and put gentle upward pressure which will detach the top plastic chamber (C) revealing a rubber gasket underneath (D). Peel off the rubber gasket gently (E).
Figure 2. Automated analysis of co-localization of host markers with mycobacterial phagosome. A. Immunostained image of Mtb infected RAW 264.7 macrophage at 24 hpi. A: Host marker (TfR) in grey and Mtb in green co-localize on phagosomes 1, 2, 3, 4 (pointed by blue arrows) to various degrees. Images have been pseudo-colored for illustration. B-H: Binary image processing to quantify co-localization as described in steps 25 to 30 of the section D of the protocol: Here, black, grey and yellow circles are symbolic representation of images C, D and F respectively. E shows pictorial representation of the effect of Boolean (XOR) operator on the images C and D image to yield F. H gives the quantification of mean fluorescence intensity (MFI) of TfR in the different ROIs with ID from 1 to 4. - Morphology operators (dilate, erode) and Boolean logic operations are integrated as processing tools in many image analysis softwares for easy use. However, they are based on complex mathematical theories and so I refer to literature (Serra, 1987; Dougherty and Lotufo, 2003) to be careful with the interpretations of these processing parameters.
- Create a binary image in the color channel of the bacteria (green). In binary images, the background will have 0 pixel intensity and the selected object (bacteria) will have a pixel intensity of 1. In the given example, Figure 2B, this can be achieved by “Thresholding” the intensity in the color channel of the bacteria. Additionally, in NIS Elements Advanced Research software, the “Autodetect all” tool can be used which works by detecting the intensity of the pixels under the cursor and so the cursor movement helps to mark all the bacteria in a given field.
Recipes
- 4% Paraformaldehyde solution in PBS (pH 7.4) (1 L)Note: Paraformaldehyde fumes are toxic. All work should be done in a ventilated fume hood.
Paraformaldehyde 40 g 10x PBS 100 ml Deionized water 800 ml - Heat 800 ml of on a hot plate to 60 °C.
- Add 40 g of paraformaldehyde while stirring on a hot plate.
- Add 50 µl of 10 N of sodium hydroxide (NaOH), continue to stir.
- Allow solution to stir until paraformaldehyde dissolves.
- Add 25 ml of 10x PBS and mix.
- Measure the pH of the solution using appropriate pH strips.
- Add water to a final volume of 1 L.
- Filter 4% paraformaldehyde through 0.45 µm filter.
- Aliquot and freeze at -20 °C for long term storage.
- Heat 800 ml of on a hot plate to 60 °C.
- DMEM complete media Filter sterilize through 0.2 µM filter and stored at 4 °C
DMEM 435 ml 1 M HEPES 10 ml 0.2 M L-glutamine 5 ml Heat inactivated fetal bovine serum 50 ml
No antibiotics should be included in the media if it is going to be used for infections - DMEM/L929 complete mediaFilter sterilize through 0.2 µM filter and stored at 4 °C
DMEM 390 ml Heat inactivated FBS 50 ml L929-cell conditioned media 50 ml 0.2 M L-glutamine 5 ml 0.1 M sodium pyruvate 5 ml
No antibiotics should be included in the media if it is going to be used for infections. - Preparation of L-Cell conditioned media
L929-cell mediumDMEM 440 ml Heat inactivated FBS 50 ml 200 mM L glutamine 5 ml 100x Non-essential amino acids 5 ml 1,000x β-mercaptoethanol 0.5 ml - Thaw 1 vial of L929 cells, add 1 ml of warm media to the vial and plate on one 15 cm TC dish in at least 35 ml of warm media.
- Allow to grow to confluence, usually takes 3-4 days when starting from frozen stock.
- Rinse the cells with 1x PBS and then cover with 10 ml 1x Trypsin EDTA and incubate at 37 °C for 1-5 min until the cells come off the bottom of the plate with gentle pipetting.
- Add an excess of L-cell media to the cells (3x the volume of Trypsin EDTA added) and pipette up and down to break clumps.
- Centrifuge at 650 x g for 5 min.
- Resuspend the cells in 25 ml of media.
- Add 1 ml of cells to each of 25 of 15 cm TC dishes containing 38 ml of growth media.
- Allow cells to grow for about 5-7 days, the cells should reach confluence on day 6. Pipet off media and filter with a 0.22 µM filter.
- Aliquot into 50 ml vials and freeze at -80 °C for use up to six months.
- Thaw 1 vial of L929 cells, add 1 ml of warm media to the vial and plate on one 15 cm TC dish in at least 35 ml of warm media.
- 2% BSA in PBS containing 0.1% saponin (50 ml)
- Weigh 0.1 g of saponin and dissolve in 100 ml of 1x PBS. Filter through 0.45 µm and stored at room temperature.
- Dissolve 2 g BSA in 50 ml of PBS-0.1% saponin gently (prepare fresh).
- Weigh 0.1 g of saponin and dissolve in 100 ml of 1x PBS. Filter through 0.45 µm and stored at room temperature.
- 2% BSA in PBS containing 0.1% triton X-100 (50 ml)
- Weigh 0.1 g of triton X-100 (w/v) and dissolve in 100 ml of 1x PBS. Filter through 0.45 µm and stored at room temperature.
- Dissolve 2 g BSA in 50 ml of PBS-0.1% triton X-100 gently (prepare fresh).
- Weigh 0.1 g of triton X-100 (w/v) and dissolve in 100 ml of 1x PBS. Filter through 0.45 µm and stored at room temperature.
- 7H9 complete media (1 L)
7H9 powder 4.7 g 50% glycerol 4.0 ml 20% Tween 80 2.5 ml Water to 900 ml - Dissolve 7H9 powder in water and add glycerol and Tween 80.
- Adjust the volume of water to give a final volume of 900 ml media.
- Add 100 ml of OADC for Mtb complete media or 100 ml of ADC for Msmeg and BCG complete media.
- Sterilize through 0.22 µM filter and store at 4 °C.
- Alternatively, autoclave after dissolving 7H9 and glycerol in about 900 ml water and then supplement with sterile solution of tween-80 and 100 ml of ADC/OADC and store at 4 °C.
- Dissolve 7H9 powder in water and add glycerol and Tween 80.
- Fixative
4% PFA/PBS for fixing BCG and Msmeg
1% PFA/PBS for fixing Mtb - Blocking solution
2% BSA in PBS containing 0.1% detergent
Acknowledgments
I thank Jennifer A. Philips for the supervision and development of this protocol. This protocol was adapted from the published work Mehra et al. (2013). The work was supported by grants and fellowships from the NIH (R01 AI087682), the Doris Duke Charitable Foundation, the Infectious Disease Society of America, the Michael Saperstein Medical Scholars Research Fund (New York University School of Medicine), Potts Memorial Foundation and the American Society of Microbiology.
References
- Banaiee, N., Jacobs, W. R., Jr. and Ernst, J. D. (2006). Regulation of Mycobacterium tuberculosis whiB3 in the mouse lung and macrophages. Infect Immun 74(11): 6449-6457.
- Delaby, C., Rondeau, C., Pouzet, C., Willemetz, A., Pilard, N., Desjardins, M. and Canonne-Hergaux, F. (2012). Subcellular localization of iron and heme metabolism related proteins at early stages of erythrophagocytosis. PLoS One 7(7): e42199.
- Deretic, V. and Fratti, R. A. (1999). Mycobacterium tuberculosis phagosome. Mol Microbiol 31(6): 1603-1609.
- Dougherty, E. R. and Lotufo, R. A. (2003). Hands-on morphological image processing. Spie Bellingham, WA.
- Fratti, R. A., Backer, J. M., Gruenberg, J., Corvera, S. and Deretic, V. (2001). Role of phosphatidylinositol 3-kinase and Rab5 effectors in phagosomal biogenesis and mycobacterial phagosome maturation arrest. J Cell Biol 154(3): 631-644.
- Goldenthal, K. L., Hedman, K., Chen, J. W., August, J. T. and Willingham, M. C. (1985). Postfixation detergent treatment for immunofluorescence suppresses localization of some integral membrane proteins. J Histochem Cytochem 33(8): 813-820.
- Hava, D. L., van der Wel, N., Cohen, N., Dascher, C. C., Houben, D., León, L., Agarwal, S., Sugita, M., van Zon, M. and Kent, S. C. (2008). Evasion of peptide, but not lipid antigen presentation, through pathogen-induced dendritic cell maturation. Proc Natl Acad Sci U S A 105(32): 11281-11286.
- Mehra, A., Zahra, A., Thompson, V., Sirisaengtaksin, N., Wells, A., Porto, M., Koster, S., Penberthy, K., Kubota, Y., Dricot, A., Rogan, D., Vidal, M., Hill, D. E., Bean, A. J. and Philips, J. A. (2013). Mycobacterium tuberculosis type VII secreted effector EsxH targets host ESCRT to impair trafficking. PLoS Pathog 9(10): e1003734.
- Mottola, G., Boucherit, N., Trouplin, V., Oury Barry, A., Soubeyran, P., Mege, J. L. and Ghigo, E. (2014). Tropheryma whipplei, the agent of whipple's disease, affects the early to late phagosome transition and survives in a Rab5- and Rab7-positive compartment. PLoS One 9(2): e89367.
- Nagabhushanam, V., Solache, A., Ting, L. M., Escaron, C. J., Zhang, J. Y. and Ernst, J. D. (2003). Innate inhibition of adaptive immunity: Mycobacterium tuberculosis-induced IL-6 inhibits macrophage responses to IFN-gamma. J Immunol 171(9): 4750-4757.
- Patiño, S., Alamo, L., Cimino, M., Casart, Y., Bartoli, F., García, M. J. and Salazar, L. (2008). Autofluorescence of mycobacteria as a tool for detection of Mycobacterium tuberculosis. J Clinical Microbiol 46(10): 3296-3302.
- Schwebach, J. R., Jacobs, W. R., Jr. and Casadevall, A. (2001). Sterilization of Mycobacterium tuberculosis Erdman samples by antimicrobial fixation in a biosafety level 3 laboratory. J Clin Microbiol 39(2): 769-771.
- Serra, J. (1987). Morphological optics. J Microsc 145(Pt 1): 1-22.
- van der Wel, N., Hava, D., Houben, D., Fluitsma, D., van Zon, M., Pierson, J., Brenner, M. and Peters, P. J. (2007). M. tuberculosis and M. leprae translocate from the phagolysosome to the cytosol in myeloid cells. Cell 129(7): 1287-1298.
- Waters, J. C. (2009). Accuracy and precision in quantitative fluorescence microscopy. J Cell Biol 185(7): 1135-1148.
Article Information
Copyright
© 2014 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Mehra, A. (2014). Phagolysosomal Trafficking Assay . Bio-protocol 4(13): e1163. DOI: 10.21769/BioProtoc.1163.
- Mehra, A., Zahra, A., Thompson, V., Sirisaengtaksin, N., Wells, A., Porto, M., Koster, S., Penberthy, K., Kubota, Y., Dricot, A., Rogan, D., Vidal, M., Hill, D. E., Bean, A. J. and Philips, J. A. (2013). Mycobacterium tuberculosis type VII secreted effector EsxH targets host ESCRT to impair trafficking. PLoS Pathog 9(10): e1003734.
Category
Immunology > Immune cell function > Macrophage
Cell Biology > Cell imaging > Fluorescence
Microbiology > Microbe-host interactions > In vitro model > Cell line
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.