- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Deflagellation and Regeneration in Chlamydomonas
Published: Vol 4, Iss 12, Jun 20, 2014 DOI: 10.21769/BioProtoc.1155 Views: 15179
Reviewed by: Ru Zhang
Original research article
The authors used this protocol in:

Jan 2013
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
Eukaryotic cilia/flagella are one of the only cellular structures that can be removed without injuring cells, can be highly purified for biochemical analysis, and, in many cells, can be completely reassembled within 90 minutes. Following amputation, the expression of many flagellar genes is up-regulated, and many are packaged and associated with intraflagellar transport (IFT) particles for transport to flagellar bases and into growing flagella. Studies of deciliation and ciliary growth provide insight to mechanisms that regulate microtubule assembly and length, mechanisms that regulate the transport of soluble cytoplasmic proteins into the ciliary compartment and their assembly into microtubules, and mechanisms that regulate trafficking of membrane proteins and lipids to the plasma membrane or to ciliary bases and their movement into and out of the cilium. These are important for motility and for signal transduction.
Deciliation methods for many cells have been developed and most require extracellular calcium ions and activation of signaling pathways that regulate microtubule severing (Quarmby, 2009). Deciliation occurs at the distal end of the basal bodies and, as soon as axonemes are severed, the membrane reseals and basal bodies begin to regenerate cilia.
Chlamydomonas is an ideal organism with which to study ciliary regeneration. Cells are easily and inexpensively cultured, flagellar amputation and regeneration is uniform in all cells in a population and growth can be assayed by observing fixed or living cells with a phase contrast microscope equipped and a 40x objective lens. Flagellar regeneration on individual living cells can be observed using paralyzed mutants immobilized in agarose. Because deflagellation leaves cells intact, the released flagella can be purified without contamination with cellular debris. The most reliable deciliation and regeneration method is the pH shock method developed by Reference 5 (also see References 4 and 11). Other methods are reviewed by Quarmby, (2009). The pH shock method is primarily used for Chlamydomonas but can be used for deciliation and regeneration of Tetrahymena cilia (Gaertig et al., 2013).
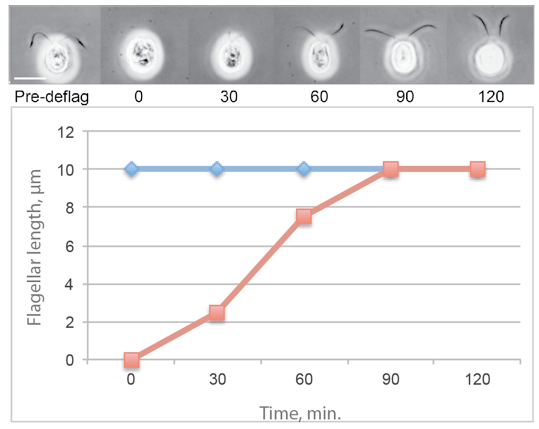
Figure 1. Typical flagellar regeneration curve showing phase contrast images of Chlamydomonas cells photographed before deflagellation and during regeneration. The average flagellar lengths of a population of regenerating cells is shown in red and the average flagellar lengths on a population of nondeflagellated cells is in blue.
Materials and Reagents
- Cells
Chlamydomonas cells can be obtained from a variety of sources and pure strains of a variety of flagellar mutants can be obtained from the Chlamydomonas Resource Center (http://chlamycollection.org/contact-us/). - Media and cell culture (see Notes)
- 0.5 N acetic acid
- 0.5 M NaOH
- 2% glutaraldehyde in M medium or Lugols iodine (see Recipes)
Note: For electron microscopy, one should use fresh glutaraldehyde from sealed ampuoles. To measure flagellar lengths, the age of the glutaraldehyde is not critical.
Optional:
- 2.5% low EEO agar (Thermo Fisher Scientific, FisherBiotechTM, catalog number: BP160-100 )
Note: It is used to observe flagellar growth or maintenance on individual living cells.
- 5 mM (final concentration) colchicine (to inhibit microtubule assembly)
Note: These experiments are carried out in phosphate-buffered Minimal medium. Avoid Tris-containing buffers because they may inhibit the effects of colchicine (Margulis et al., 1969).
- 10 µg/ml (final concentration) Cycloheximide
Note: It is used to inhibit protein synthesis.
- VALAP (see Recipes)
Note: It is used to support coverslips to examine living cells without inducing deflagellation by coverslip pressure.
Equipment
- pH meter calibrated for pH 4-7
Note: Some gel-filled electrodes are not accurate across this pH range.
- Centrifuge and tubes
Note: For small scales, a clinical centrifuge and conical tubes.
- Phase contrast microscope or brightfield microscope
Note: If cells are fixed with Lugol’s iodine, a 40x lens is ideal for flagellar length measurements.
- Orbital shaker
- Magnetic stirrer
- Stir bar
Software
- Image J (http://imagej.nih.gov/ij/)
Procedure
- Check cells with a 40x lens and a phase microscope to be certain that cells are healthy, contain two flagella, and are motile (unless paralyzed mutants are studied). Dividing cells lack flagella and are surrounded by a refractile cell wall containing 4-8 cells. Cultures with dividing cells are not synchronous and are not used for regeneration experiments.
- Harvest cells (~5x105 cells/ml) using a clinical centrifuge, for up to 120 ml of culture, or low speed preparative centrifuge (3 min, 1,500 x g) pour off the supernatant.
- Suspend cells in 20-100 ml of fresh medium in a beaker by gentle swirling or gently drawing cells up and down through a large-bore pipette (I break off the tips of plastic pipettes.).
- Check cells with a phase microscope to insure that they are uniformly flagellated and have not been deflagellated during centrifugation and resuspension.
Optional: Agarose to observe flagella on immobilized living cells.- Dissolve 2.5% low EEO agar in culture medium and maintain at 45 °C for use.
- Apply cells to a clean coverslip, draw off most of the liquid, and apply ~20 microliters of agarose at 40 °C.
- Rapidly invert over a clean microscope slide and seal the edges with VALAP (1:1:1 Vaseline, Lanolin, Paraffin).
- Flatten the bead of VALAP around the edge of the coverslip with a warm spatula.
Note : To capture time-lapse images of regenerating flagella use a microscope equipped with a UV filter. Keep the illumination low and be certain that non-deflagellated cells maintain full length throughout the observation period (see Reference 1).
- Dissolve 2.5% low EEO agar in culture medium and maintain at 45 °C for use.
- Stir cells using a magnetic stirrer and dropwise add 0.5 N acetic acid until the pH reaches 4.5-4.0. Add acetic acid as rapidly as possible but be certain that it is well mixed as it is added to the medium. Keep at pH 4 for no longer than ~1 minute and then raise the pH to 6.8-7.2 by dropwise adding 0.5 N KOH. For 100 ml of cells in M medium, use approximately 4.5 ml of 0.5 N acetic acid and 5.2 ml of 0.5 M KOH. For most purposes, it is best to monitor the pH change but, for mass-deflagellations in microtitre plates, adding defined quantities of acetic acid and KOH is useful.
- For regeneration
- Pellet cells using a clinical centrifuge, and suspend in fresh M medium to ~105 cells/ml. It is not critical to suspend in fresh medium but I generally do so.
- Incubate cells in light with aeration using bubblers or by gentle rotation in an Erlenmeyer flask on an orbital shaker.
- Flagella generally will start to grow within 10-15 min and will be fully grown by 60-90 min.
- To measure flagella on regenerating cells, fix samples at 5-10 min intervals with glutaraldehyde or Lugol’s iodine.
- Fix: 2% glutaraldehyde in M medium. Dilute 100 microliters of cells + 100 microliters of 2% glutaraldehyde and let settle in microfuge tubes. Measure flagella within 1-2 days. Longer fixation will lead to clumping of cells at the bottom of the tube, which makes measurement of individual flagella very difficult.
- Alternatively, fix by diluting 80 microliters of cells + 20 microliters Lugol’s iodine. Measure flagella within 1-2 days to avoid clumping of flaglellated cells.
- Regeneration can be inhibited reversibly by the addition of colchicine, an inhibitor of microtubule assembly. If cycloheximide is added at the time of deflagellation, flagellar regeneration will be limited to half-length due to the exhaustion of a pool of proteins critical for flagellar assembly (Rosenbaum et al., 1969).
- Fix: 2% glutaraldehyde in M medium. Dilute 100 microliters of cells + 100 microliters of 2% glutaraldehyde and let settle in microfuge tubes. Measure flagella within 1-2 days. Longer fixation will lead to clumping of cells at the bottom of the tube, which makes measurement of individual flagella very difficult.
- Pellet cells using a clinical centrifuge, and suspend in fresh M medium to ~105 cells/ml. It is not critical to suspend in fresh medium but I generally do so.
- Measuring flagella: There are a variety of ways to measure flagella and one has to balance accuracy with the ease of measuring large numbers of flagella to minimize error. Even if flagella are planar when fixed, they are curved and are viewed at various angles, depending on the orientation of the cell on the microscope slide. Very short flagella may be difficult to see if the cell is not properly oriented to reveal the flagella. It also is difficult to identify the flagellar base because it is surrounded by the cell wall, which can obscure the first micron of a flagellum above the basal body. Thus, the observer must use their judgment when measuring flagella and be consistent with all measurements. One can use complex image analysis involving the Z-axis (see Reference 9) but one also could simply deflagellate cells at various time points, photograph isolated flagella attached to a coverslip, and measure their lengths with a variety of image programs.
The easiest method to measure flagella is to use an ocular micrometer and record measurements of flagella on fixed cells by hand. I prefer to use a ccd or video camera that interacts with Image J, a free software program (http://imagej.nih.gov/ij/). Trace the flagella using a mouse, save the measurements, and determine the average lengths and distributions of length (using histograms) using Excel or similar spreadsheets.
- Flagella are easily purified from pH-shocked cells following methods described elsewhere (these protocols).
Notes
- Notes about media and cell culture
- Culture media recipes are available at http://www.chlamy.org/media.html.
- I prefer to culture cells in at room temperature in Sager and Granick (1953) minimal (M) medium (http://www.chlamy.org/SG.html; Harris, 1989) and synchronize cells on a 12 h dark/light cycle to insure that all cells are at the same point in the cell cycle. This is important because Chlamydomonas cells disassemble their flagella prior to division and one mutant, pf18, gradually disassembles its flagella 4-6 h after the beginning of the light cycle (Tuxhorn et al., 1998). Cell density is not critical but cells grown to stationary stage (> 106 cells/ml) often are not 100% flagellated.
- Autoclave media in Erlenmeyer flasks fitted with a foam plug and cotton-plugged glass Pasteur pipette for aeration with house air or an aquarium pump. For deflagellation and regeneration, cultures of 50-100 ml of cells grown to ~ 5 x 105 cells/ml is adequate.
- To isolate flagella, culture 8-16 L of cells in aerated 4 L bottles and concentrate cells to 100-200 ml for pH shock.
- Culture media recipes are available at http://www.chlamy.org/media.html.
Recipes
- Lugol’s Iodine
Mix 6% potassium iodide in distilled water and add 4% iodine crystals
Mix by stirring overnight and store indefinitely
Acknowledgments
This protocol was adapted from Lefebvre (1995), Margulis et al. (1969) and Witman (1986).
References
- Dentler, W. L. and Adams, C. (1992). Flagellar microtubule dynamics in Chlamydomonas: cytochalasin D induces periods of microtubule shortening and elongation; and colchicine induces disassembly of the distal, but not proximal, half of the flagellum. J Cell Biol 117(6): 1289-1298.
- Gaertig, J., Wloga, D., Vasudevan, K. K., Guha, M. and Dentler, W. (2013). Discovery and functional evaluation of ciliary proteins in Tetrahymena thermophila. Methods Enzymol 525: 265-284.
- Harris, E. H. (1989). A comprehensive guide to biology and laboratory use. The Chlamydomonas Sourcebook. Academic Press
- Lefebvre, P. A. (1995). Flagellar amputation and regeneration in Chlamydomonas. Methods Cell Biol 47: 3-7.
- Margulis, L., Banerjee, S. and White, T. (1969). Colchicine-inhibited cilia regeneration: explanation for lack of effect in tris buffer medium. Science 164(3884): 1177-1178.
- Quarmby, L. M. (2009). Deflagellation. In: Witman, G. B. (ed). The Chlamydomonas Sourcebook second edition. Academic Press Vol 3:43-69.
- Rosenbaum, J. L., Moulder, J. E. and Ringo, D. L. (1969). Flagellar elongation and shortening in Chlamydomonas. The use of cycloheximide and colchicine to study the synthesis and assembly of flagellar proteins. J Cell Biol 41(2): 600-619.
- Sager, R. and Granick, S. (1953). Nutritional studies with Chlamydomonas reinhardi. Ann N Y Acad Sci 56(5): 831-838.
- Saggese, T., Young, A. A., Huang, C., Braeckmans, K. and McGlashan, S. R. (2012). Development of a method for the measurement of primary cilia length in 3D. Cilia 1(1): 11.
- Tuxhorn, J., Daise, T. and Dentler, W. L. (1998). Regulation of flagellar length in Chlamydomonas. Cell Motil Cytoskeleton 40(2): 133-146.
- Witman, G. B. (1986). Isolation of Chlamydomonas flagella and flagellar axonemes. Methods Enzymol 134: 280-290.
Article Information
Copyright
© 2014 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Dentler, W. (2014). Deflagellation and Regeneration in Chlamydomonas. Bio-protocol 4(12): e1155. DOI: 10.21769/BioProtoc.1155.
Category
Plant Science > Phycology > Cell analysis
Cell Biology > Cell movement > Cell motility
Cell Biology > Cell imaging > Live-cell imaging
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.