- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Quantification of T Cell Antigen-specific Memory Responses in Rhesus Macaques, Using Cytokine Flow Cytometry (CFC, also Known as ICS and ICCS): from Assay Set-up to Data Acquisition

Published: Vol 4, Iss 8, Apr 20, 2014 DOI: 10.21769/BioProtoc.1110 Views: 17994
Reviewed by: Jia LiAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Nov 2012
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
What was initially termed ‘CFC’ (Cytokine Flow Cytometry’) is now more commonly known as ‘ICS’ (Intra Cellular Staining), or less commonly as ‘ICCS’ (Intra Cellular Cytokine Staining). The key innovations were use of an effective permeant (allowing intracellular staining), and a reagent to disrupt secretion (trapping cytokines, thereby enabling accumulation of detectable intracellular signal). Because not all researchers who use the technique are interested in cytokines, the ‘ICS’ term has gained favor, though ‘CFC’ will be used here.
CFC is a test of cell function, exposing lymphocytes to antigen in culture, then measuring any cytokine responses elicited. Test cultures are processed so as to stain cells with monoclonal antibodies tagged with fluorescent markers, and to chemically fix the cells and decontaminate the samples, using paraformaldehyde.
CFC provides the powers of flow cytometry, which includes bulk sampling and multi-parametric cross-correlation, to the analysis of antigen-specific memory responses. A researcher using CFC is able to phenotypically characterize cells cultured with test antigen, and for phenotypic subsets (e.g. CD4+ or CD8+ T cells) determine the % frequency producing cytokine above background level.
In contrast to ELISPOT and Luminex methods, CFC can correlate production of multiple cytokines from particular, phenotypically-characterized cells. The CFC assay is useful for detecting that an individual has had an antigen exposure (as in population screenings), or for following the emergence and persistence of antigen memories (as in studies of vaccination, infections, or pathogenesis). In addition to quantifying the % frequency of antigen-responding cells, mean fluorescence intensity can be used to assess how much of a cytokine is generated within responding cells.
With the technological advance of flow cytometry, a current user of CFC often has access to 11 fluorescent channels (or even 18), making it possible to either highly-characterize the phenotypes of antigen-responding cells, or else simultaneously quantify the responses according to many cytokines or activation markers. Powerful software like FlowJo (TreeStar) and SPICE (NIAID) can be used to analyse the data, and to do sophisticated multivariate analysis of cytokine responses.
The method described here is customized for cells from Rhesus macaque monkeys, and the extensive annotating notes represent a decade of accumulated technical experience. The same scheme is readily applicable to other mammalian cells (e.g. human or mouse), though the exact antibody clones will differ according to host system. The basic method described here incubates 1 x 106 Lymphocytes in 1 ml tube culture with antigen and co-stimulatory antibodies in the presence of Brefeldin A, prior to staining and fixation.
[Historical Background] The first report of fixing and permeabilizing lymphocytes, then staining them with antibodies against IFN gamma, was made by Andersson et al. in 1989. In 1991, Sander et al. demonstrated improved methods, using paraformaldehyde to fix cells, saponin (an amphipathic glycoside) to permeabilize them, and fluorescently-labeled antibodies to stain intracellular cytokines for microscope examination. In 1993, Jung et al. extended this method for use with flow cytometry, included monensin (a polyether antibiotic ionophore which blocks intracellular protein transport) to inhibit secretion, so as to increase the intracellular signal of the cytokine molecules that would otherwise be released soon after synthesis. In 1995, Prussin and Metcalfe used directly-conjugated antibodies, and reported good results with 6 h incubations. Also in 1995, Picker et al. considerably enhanced the sensitivity and reproducibility of cytokine detection by using Brefeldin A (‘BfA’, a fungal lactone antibiotic) to block the cytokine-secretion apparatus, and by using a different permeant (Tween-20). This improved method was applied by Picker et al. in a 1997 report of the antigen-specific homeostatic mechanism in human HIV+ patients. In 2001, Schuerwegh et al. confirmed that BfA provides better cytokine signal in the assay than does monensin, though monensin is still used widely by others in this method.
Regarding non-human primate studies, two reports in 1989, one by Gardner and another by McClure, showed that Rhesus macaques were a useful model for studying HIV disease and AIDS. In 2002, Picker et al. reported the application of a specially-modified CFC assay to Rhesus macaques. In 2012, a consortium-appointed group aiming to establish standards for collaborating groups using CFC in Rhesus vaccine studies published their recommendations for a 96-well plate method with a 6 h total incubation (Donaldson et al., 2012; Foulds et al., 2012).
The general procedure reported here is that 2002 tube-format (see Note 1) method, now with a 9 h total incubation, and optimized especially for low-end sensitivity. The specific details here are the state of the art now practiced by the Picker Lab, at the Oregon Health and Science University, affiliated with the Oregon National Primate Research Center. These methods have been used in several of our recent publications (Hansen et al., 2013a; Hansen et al., 2013b, Fukazawa et al., 2012; Hansen et al., 2011; Hansen et al., 2009). It is important to note that in our hands, plate-format CFC is not as sensitive and reproducible for weak responses as is this tube-based method described here (unpublished observations). Until that difference is understood and solved, the tube-based method remains the most-sensitive format for CFC.
Materials and Reagents
- Lymphocyte suspension from Rhesus macaques blood (Note 2) or bronchoaveolar lavage (BAL), or harvest from solid biopsy or necropsy tissue, cell density determined by a method accurate for the sample (Notes 3 and 4)
- Need ~1 x 106 viable lymphocytes per test
- Freshly-obtained (Note 5)
- OR thawed cryopreserved sample (Note 6)
- Need ~1 x 106 viable lymphocytes per test
- Antigen
- Negative control(s) (Note 9)
- Positive control:
Superantigen Staphylococcus Enterotoxin B SEB (Note 10) (Toxin Technology, catalog number: BT202 ) (lyophilized powder, 100 μg; stock: 100 μg/ml in water; usage: 2 μl/test) - Other positive control (experiment-specific)
- Peptide mixes (1-100 different peptides, at ≥ 2 μg/peptide/1 ml-test) 15 amino acid peptides (15 mers) overlapping by 11 amino acids
- Negative control(s) (Note 9)
- Antibody
- Unconjugated antibody for costimulation during culture incubation (Note 11)
Anti-CD28, pure unconjugated, clone CD29.2 (Note 12)
Anti-CD49d, pure unconjugated, clone 9F10
Stocks diluted to 0.5 mg/ml; use 1 μl per 1 x 106 Ly - Essential fluorophore-conjugated monoclonal antibodies
The fluorophores you use are dependent upon the flow cytometer available to you. Many companies sell the appropriate fluorophore-conjugated antibodies, including BD, Beckman Coulter, Life Technologies, InvitrogenTM, eBiosciences, and many others.
Anti-CD3e, clones reactive with Rhesus (SP34-2, FN18)
Anti-CD4 (L200, MT477))
Anti-CD8a (SK1, RPA-T8)
Anti-CD69 (FN50, CH/4, TP1.55.3) (Note 13)
Anti-IFNg (B27)
Anti-TNFa (MAB11) - Optional fluorophore-conjugated monoclonal antibodies
Anti-CD45 (DO58-1283) (Note 14)
Anti-IL2 (MQ1-17H12)
Anti-MIP1b (D21-1351)
Ant-CD107 (alpha: H4A3, beta: H4B4) (Note 15)
Anti-CD95 (DX2)
Anti- CD45RA (L48, 5H9, MEM-56, others) (Note 41)
Anti-CCR7 (CD197) (Note 42)
Anti-Ki67 (B56) (Note 38)
- Unconjugated antibody for costimulation during culture incubation (Note 11)
- Brefeldin A (Sigma-Aldrich, catalog number: B-7651 )
Vendors: (Sigma-Aldrich, catalog number: B-7651; BioLegend, catalog number: 91850 )
Working stock: 10 mg/ml, in DMSO (1.0 μl/test) (Note 16) - Benzonase (Merck KgaA, Novagen) (use at 50 U/ml)
- 1x RPMI-1640 (w/o L-glutamine, 0.1 μm filtered) (e.g. HyCone, catalog number: SH30096.02 )
- Fetal Bovine Serum (FBS, aka: 'FCS') (e.g. HyClone, catalog number: SH30070.03 ) (defined, heat-inactivated, 40 nm-filtered)
- Penicillin+Streptomycin (P/S) Solution (e.g. Sigma-Aldrich, catalog number: P-0781 )
- L-glutamine (200 mM) (e.g. Sigma-Aldrich, catalog number: G-7513 (100 ml)
- Sodium pyruvate (SP) (e.g. Sigma-Aldrich, catalog number: S-8636 ) (100 ml)
- Beta-Mercaptoethanol (bME) (e.g. Sigma-Aldrich, catalog number: M-7522 ) (100 ml) (Note 40)
- Sterile-filtration apparatus (e.g. Corning, catalog number: 430769 ) (500 ml capacity 0.22 μm cellulose-acetate filter)
- Dulbecco's Phosphate Buffered Saline (DPBS) (e.g. Thermo Fisher Scientific, Corning, catalog number: 55-031-PB )
- Bovine serum albumin (BSA) (e.g. Thermo Fisher Scientific, catalog number: BP1605100 )
- Sodium azide (preservative; NaN3) (e.g. Thermo Fisher Scientific, catalog number: BP922-500 )
- BD FACS Lysing Solution (10x concentrate) (BD, catalog number: 349202 )
- Tween-20 (polyoxyethylenesorbitan monolaurate) (e.g. Sigma-Aldrich, catalog number: P-7949 )
- Aqua LIVE/DEAD kit (Life Technologies, InvitrogenTM, www.lifetechnologies.com, search 'LIVE/DEAD' for an evolving array of stains and kits) (Note 43)
- Concentrated dye stock (see Recipes)
- Staining Solution (made fresh) (see Recipes)
- Concentrated dye stock (see Recipes)
- Tissue culture medium ('R10') (see Recipes)
- 'PAB' wash buffer (see Recipes)
- 'Lyse' fixation and RBC-lysing solution (see Recipes) (Note 17)
- 'Perm' fixation and cell-permeabilizing solution (see Recipes) (Note 18)
Equipment
- Tubes (Notes 1 and 7) polypropylene (PP) (round-bottom, 5 ml/12 x 75 mm, sterile) (e.g. Falcon®, catalog number: 35-2054 )
- Computer-generated printed labels for tubes (optional; must stick well to polypropylene)
- Tube-holding racks (optional) (e.g. Thermo Fisher Scientific, No-Wire Grip Rack 10-13 MM 90 place) (Note 8)
- Foam cosmetic wedges (or functional equivalent) (The ones we use are 2" long x ¾" high when lying on the long side.)
- Laminar flow biosafety cabinet (for sterility, even if not working with pathogens)
- Trapped vacuum aspirator
- Appropriate fluid measuring dispensers, with appropriate disposables
- Electric pump pipettors for measurements between 1-50 ml
- Manual hand micropipettors for measurements between 0.5-1,000 μl
- Repeater-dispensers (e.g. from Eppendorf)
- Hand repeaters, for measurements between 0.5-2 ml
- Stationary pump dispensers, for measurements between 1-5 ml
- Electric pump pipettors for measurements between 1-50 ml
- Centrifuge with swing-buckets (capable of 800 x g) (e.g. Sorvall Legend T/RT)
- Vortexer
- Lab timer
- Incubator for tissue culture (humidified, stable at 37 °C, 5% CO2 atmosphere)
Option: Standard water-jacketed T/C incubator (e.g. Thermo Fisher Scientific, FormaTM, Series II, Model: 3110 )
Option: UniBator (Tritech Research) DigiTherm CO2 incubator with rapid cooling and bi-directional interface (Note 19) - Refrigerator at 4 °C
- Flow cytometry analyser, 6-fluorescence detectors or more (e.g. BD, model: LSR-II )
- A method of counting PBMC (e.g., Coulter counter, Guava, or hemocytometer)
Procedure
Note: Please see Figures 1 and 2 for the flow chart of the CFC set up process and the CFC tube staining process, respectively. 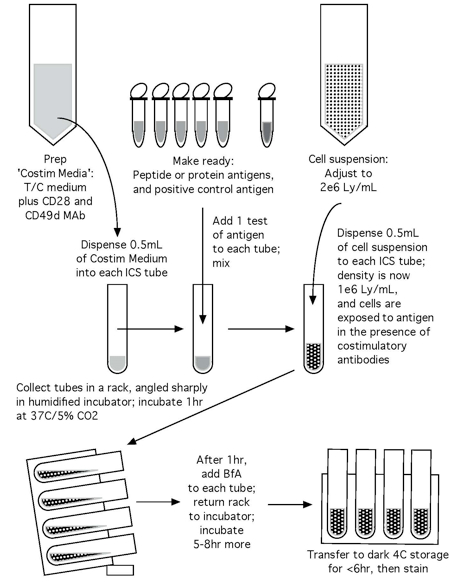
Figure 1. Flow chart of the CFC set up process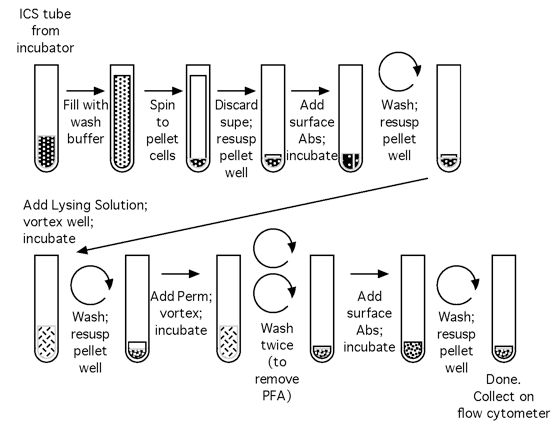
Figure 2. Flow chart of the CFC tube staining process
Part I. Culture setup, incubation, chilling
- Pre-start preparation
- See QA/QC notes.
- Make fresh medium (or begin warming to room temperature).
- Label PP tubes.
- Begin thawing peptides in DMSO (if applicable).
- See QA/QC notes.
- Obtain cells
- Freshly-harvested WBC (Notes 2-5).
- PBMC (peripheral blood mononuclear cells) from blood. (Other protocols explain how to isolate PBMC from blood. It is also possible to use whole blood that has been treated with a hypotonic saline like ACK to burst red blood cells.)
- BAL (bronchoaveolar lavage, sieved).
- Solid tissue, mechanically processed (LN, spleen, bone).
- Solid tissue, enzymatically processed (gut, liver, vagina).
- Cryopreserved WBC harvests.
- Thaw (Note 20).
- Rest (Note 20).
- Determine viability (Note 20).
- Thaw (Note 20).
- Freshly-harvested WBC (Notes 2-5).
- Adjust suspension density to approximately 2 x 106 Ly/ml, in fresh-made R10 (Note 21)
- e.g. 1 x 106 Ly/500 μ.
- R10 un-supplemented by antibodies or antige.
- Set suspension aside at room temperature until use.
- e.g. 1 x 106 Ly/500 μ.
- Prepare R10 supplemented with costimulating MAb anti-CD28, anti-CD49d (Note 11)
- Determine the total volume needed for the assay setup: (# tubes) x (500 μl/tube) x 1.2 (i.e., increase by 20%).
- Measure R10 into vessel labeled 'costim-R10'.
- Add anti-CD28 and anti-CD49d:
- Presuming [stock] = 0.5 mg/ml.
- Use 1 μl/500 μl (i.e., 2 μl/ml R10).
- Mix (vortex or inversion).
- Presuming [stock] = 0.5 mg/ml.
- Determine the total volume needed for the assay setup: (# tubes) x (500 μl/tube) x 1.2 (i.e., increase by 20%).
- Prepare substocks of R10 + costim, supplemented with antigens
- Determine the total volume needed for a particular antigen (# tubes) x (500 μl/tube) x 1.1 (i.e., increase by 10%).
- Measure R10 + costim into vessel labeled by antigen.
- Briefly vortex the stock antigen is mixed and homogenous.
- Ensure that the measurement is accurate (Note 22).
- Mix (vortex or inversion).
- Briefly vortex the stock antigen is mixed and homogenous.
- Determine the total volume needed for a particular antigen (# tubes) x (500 μl/tube) x 1.1 (i.e., increase by 10%).
- Dispense 500 μl of R10 + costim + Ag into all appropriately-labeled tubes
- Use the tube labels to consistently orient the tubes in a reproducible way in the holding rack(s).
- Vortex the solution immediately before using (it is convenient to use a repeater for this dispensing).
- Dispense the 500 μl/tube, aiming down to a particular 'side' the tube (that side is now a possible cause of cross-contamination when using a repeater, so subsequent steps should orient the tubes differently, to avoid cross-contamination).
- Put the tube-holding rack(s) in a 37 °C, 5% CO2 incubator until use (these tubes can be prepared up to 2 h before adding cells).
- Use the tube labels to consistently orient the tubes in a reproducible way in the holding rack(s).
- Dispense 500 μl of cell suspension into all racked tubes
- Be attentive to the label orientation on the tubes; orient the tubes such that dispensed suspension goes down an opposite side of the tubes (to minimize the risk of cross-contamination).
- It is convenient to use a repeater for this step.
- Always dispense into 'Negative' control tubes first.
- Always dispense into 'SEB' positive control tubes last.
- This step should take place as quickly as possible, since it is combining cells with antigen. At room temperature, however, the cells are not yet very metabolically active.
- It is not necessary to individually vortex tubes after this combination.
- Be attentive to the label orientation on the tubes; orient the tubes such that dispensed suspension goes down an opposite side of the tubes (to minimize the risk of cross-contamination).
- Transfer the racks to an incubator stably at 37 °C, 5% CO2
- Ensure that a humidifying tray contains water (or else evaporation from the tubes will concentrate cells, Ag, and salts, changing the assay outcome).
- Place the rack on its side, resting the rack top on a cosmetic wedge. The sharp angling of the tubes makes the medium more shallow, making gas diffusion easier. This orientation also makes it so that the cells settle over a larger area, with less cell-cell contact. This seems to improve response sensitivity, for reasons that remain unclear.
- Set a timer for 1 h.
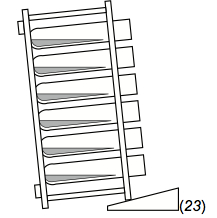
Figure 3. How to angle a rack of ICS tubes during incubation
- Ensure that a humidifying tray contains water (or else evaporation from the tubes will concentrate cells, Ag, and salts, changing the assay outcome).
- After 1 h of incubation,add Brefeldin A to all tubes (Note 24)
- Prepare a dilute cocktail, to be used at 50 μl/tube.
- If (BfA stock) is 10 mg/ml, use 1 μl/tube.
- Dilute the BfA into unsupplemented R10. 1 part BfA into 39 parts R10; deliver 40 μl of this mix into each ICS tube. A 40 μl drop will reliably slide down the wall of a PE tube.
- If (BfA stock) is 10 mg/ml, use 1 μl/tube.
- Using a repeater, shoot the BfA downward into the tube.
- Orient the labels to minimize cross-contamination.
- Dispense in a way to minimize the disruption of the settled cell pellets in each tube.
- 40 μl produces a heavy enough drop toreliably run down the side of the tube.
- Orient the labels to minimize cross-contamination.
- Do not vortex or further mix the tube contents.
- The tubes should be uncapped (or with loosely-applied 'snap caps'. It is important that easy gas exchange is allowed. The pen/strep is typically very effective at preventing bacterial and fungal growth during the short incubation period.
- Prepare a dilute cocktail, to be used at 50 μl/tube.
- Return the racks to the incubator (humidified, 37 °C, 5% CO2)
- Incubate tubes for an additional 5-8 h (with rack angled as in Figure 3) (Note 25).
- At the end of the incubation, the racks should be cooled
- The simplest way to accomplish this is to transfer the racks to a refrigerator at 4 °C.
- Tritech Research sells novel incubators that can, by computer-clock control, cool rapidly (37 °C to 15 °C, in less than 20 min; eventually to 4 °C) (Note 19).
- The simplest way to accomplish this is to transfer the racks to a refrigerator at 4 °C.
Part II. Staining
- Samples should be processed to fixation within 12 h
- Although the mammalian cells are metabolically nearly inert at 4 °C, they remain alive. Left too long, the CFC signal will change.
- Transfer the culture tubes from the incubation racks to centrifuge bucket inserts.
- Although the mammalian cells are metabolically nearly inert at 4 °C, they remain alive. Left too long, the CFC signal will change.
- Wash
- Using a bottletop pump dispenser, fill all tubes with PAB wash buffer.
- Centrifuge 800 x g, 10 min (sufficient to produce a tight cell pellet in a 5 ml tube). The temperature of the run doesn't matter much, so long as it is well below metabolically-active 37 °C; the cells will soon be fixed.
- Remove supernatant (Note 26)
- Method 1: Wand vacuum-aspiration (Note 27).
- Method 2: Wand vacuum-aspiration with stop (Note 28).
- Method 3: Decanting (Note 29).
- Method 4: Decanting with insert gasket (Note 30).
- Method 1: Wand vacuum-aspiration (Note 27).
- Resuspend pellets (Video 1) (Note 31).
 Video 1. Hand-decanting wash supernatant from polypropylene tubes used in CFC (ICS) assay
Video 1. Hand-decanting wash supernatant from polypropylene tubes used in CFC (ICS) assay
- Using a bottletop pump dispenser, fill all tubes with PAB wash buffer.
- Stain with Aqua LIVE/DEAD discriminator dye (optional, esp. for thawed cells)
- Wash the cells with ice-cold 1x PBS.
- Aspirate to remove supernatant (leaving minimal residual fluid).
- Resuspend cells (as in Part II, step B4) in 100 μl ice-cold 'Staining Solution' (100 μl/1 x 106 Ly).
- Incubate 20 min in dark, on ice.
- Wash (see Part II-B) to stop.
- Wash the cells with ice-cold 1x PBS.
- Apply surface antibody cocktail (Note 32)
- Cocktail can be made hours ahead of time, in PAB.
- Formulate cocktail at 50 μl/tube Example: CD45, CD4 (Note 44).
- Dispense with a repeater.
- Vortex the tubes after adding the cocktail to the tubes.
- Incubate at room temperature for 30 min (in the dark) (Note 33).
- If the surface stain includes a two-stage biotin-streptavidin stain, the first cocktail should include the biotinylated reagent. After the 30 min incubation, add the streptavidin reagent to the tubes (no pre-washing is needed). It is convenient to put the streptavidin in a 50 μl/tube PAB dilution, which allows for repeater dispensing.
- Cocktail can be made hours ahead of time, in PAB.
- Wash (see Part II-B)
- Apply 1 ml 'Lyse' per tube (Notes 17 and 34)
- Set a lab timer for 10 min.
- Ensure all the pellets are effectively disrupted.
- Have a plan figured out that will allow vortexing within two minutes of when you start to dispense Lyse, and that ensures the tubes get vortexed in the same order the Lyse was added.
- Fill an appropriately-sized repeater.
- Dispense quickly into tubes.
- Start the timer before adding to the first tube.
- Aim the repeater towards the midpoint wall.
- Start the timer before adding to the first tube.
- Begin vortexing tubes within 2 min of receiving Lyse.
- How Lyse is added is one of the most important details in this entire process. If the cells are not well-resuspended prior to adding Lyse, they will be fixed together, degrading data quality. If you do not mix the cells and Lyse quickly after combining them, you might get poor rupture of RBCs, degrading data quality. It is important to vortex soon and vigorously after adding the Lyse.
- If time allows, vortex the tubes again (this is especially valuable if the samples contain RBC contamination).
- How Lyse is added is one of the most important details in this entire process. If the cells are not well-resuspended prior to adding Lyse, they will be fixed together, degrading data quality. If you do not mix the cells and Lyse quickly after combining them, you might get poor rupture of RBCs, degrading data quality. It is important to vortex soon and vigorously after adding the Lyse.
- Incubate ten full minutes at room temperature, in the dark.
- Set a lab timer for 10 min.
- Wash (see Part II-B)
Adding PAB reduces the hypotonicity of the Lyse reagent, slowing the osmotic swelling of the cells, and diluting the paraformaldehyde chemically fixing the cellular proteins. The residual fluids will match those dilutions (Note 35). - Apply 0.5 ml 'Perm' per tube (Note 18)
Basically, repeat the same method used for 'Lyse', but with 0.5 ml/tube. - Wash (Note 36)
- Wash (Note 37)
- Apply intracellular antibody cocktail (Note 32)
- Cocktail can be made hours ahead of time, in PAB.
- Formulate cocktail at 50 μl/tube Example: CD3, CD8, CD69, cytokines (Note 44).
- Dispense with a repeater.
- Vortex the tubes after adding the cocktail to the tubes.
- Incubate at room temperature for 30-45 min (at room temperature, in the dark) (Note 33).
If the cocktail includes Ki67, the incubation should be lengthened to 45 min (Note 38).
- Cocktail can be made hours ahead of time, in PAB.
- Wash (Note 39)
- Remove as much fluid as possible, to ensure fast acquisition times.
- Remove as much fluid as possible, to ensure fast acquisition times.
Part III. Data Acquisition
- See Notes about QA/QC.
- Samples should be acquired within 72 h (Notes 46 and 47).
Notes
- Presented here is exclusively the tube-format method. While it is true that plate-based methods provide many advantages (multi-channel processing, smaller reagent volumes, streamlined process, robotic acquisition), we learned the hard way that some aspect of that method results in a loss of low-end sensitivity to test antigen. Whereas both the tube and plate method yield comparable CFC results above approximately 0.2% cytokine response, the tube method in our hands consistently performs better with weak responses below that level. We recognized this by 2007, and have been unable yet to modify a 96-well plate method to achieve the same low-end results.
- When using blood, an anticoagulant must be used, otherwise the cells of interest will be lost in clot formation. The Picker Lab prefers ACD (acid-citrate-dextrose, 'sodium citrate') as an anticoagulant. Repeated assessments have demonstrated (unpublished observtions) that CFC results are qualitatively different, and quantitatively less, when heparin is used. In our work, virus released to the extracellular fluid is quantified using PCR, so the anticoagulant chelating agent EDTA cannot be used. Otherwise, it has been demonstrated to work in CFC as well as ACD.
- Coulter counters (e.g. the Beckman Coulter Ac*T diff5 noted in the Equipment section) are designed to quantify human cells in blood, though they can be calibrated for Rhesus cells. The technology, however is not designed for samples containing significant non- or sub-cellular debris, as can be plentiful in cryopreserved samples recovered from a thaw, or from solid tissue biopsy mechanically and/or enzymatically treated to harvest lymphocytes. In any case not starting with fresh blood, a Coulter counter will report cell density inaccurately, usually by overcounting. Moreover, this kind of counter cannot reliably distinguish between living and dead cells. In our careful (unpublished) examinations of this issue, we found that these counters over-count lymphocyte harvests from lymph node and spleen, typically by a factor ranging from 1-2 fold. Lymphocyte harvests from lung wash or resulting from enzymatically-digested tissues (e.g. liver, gut) are functionally uncountable with a counter based on this technology because of wildly-variable amounts of debris that can populate the counting gates. And in cases of samples undergoing freeze-thaw, the viability cannot be assessed reliably. In these cases, other technologies must either augment the counts (e.g. a viability stain), or replace the Coulter counter technology. One widely-used alternative is a Guava counter, using one pass with a live-dead stain (e.g. ViaCount), and another pass gating by at least scatterplot and CD3+. This can be expensive and time-consuming. It doesn't matter exactly how one counts the cells for this assay, but users should be aware that most counting methods (even hemocytometers) are inaccurate for clinical non-blood WBC. Since all these samples will be collected on a flow cytometer, which is an instrument capable of reporting a precise number regarding the number of T cells assayed, the assay itself provides a means to QC the counting method used for input.
- The CFC method described in this protocol presumes an input of 1 x 106 lymphocytes per test, and antibody and reagent inputs are meant to be proportional. In practice, however, it has been difficult to correctly pre-quantify lymphocytes obtained from bronchoaveolar lavage (BAL) or from the hours-long enzymatic harvests from gut or liver tissue, so empirically-determined approximations are used. We have found (unpublished) that in 'dirty' cell suspension inputs (e.g. BAL, enzymatic digestions) that the strict proportionality of lymphocytes to lymphocyte-marking antibodies breaks down. For example, if a Coulter count reports that a BAL input into a CFC tube is 1e6 lymphocytes, but in actuality the input was 1/20 of that, we have found that using 1/20 of the staining antibodies yields qualitatively and quantitatively worse results than just presuming that the input was actually 1e6 lymphocytes. We hypothesize that the explanation is that 'dirty' samples have plentiful low-affinity non-specific binding sites, which 'sponge-up' much of the antibody reagent. Therefore, the CFC method described here still works effectively with 'dirty' samples, even when the input lymphocytes are far below the 1e6 target. The staining doesn't manifest as significant over-titering, and it's not possible to save money by reducing input reagents, because of the need to saturate non-specific binding sites.
- We routinely let fresh blood samples sit variable times (1-6 h) at room temperature, before processing, without noticeable effect on our CFC results. We have been forced by occasional circumstance to use blood left overnight (or shipped overnight), and have noticed a variable qualitative and quantitative loss in signal quality.
- We have found (unpublished) that PBMC samples that undergo cryopreservation then freezing results in a enrichment of memory phenotypes, and a relative loss of naïve cells. This skewing is variable from sample to sample, and so not easily corrected. It is a significant reason we do nearly all our CFC using freshly-harvested lymphocyte preparations. Other practitioners may not have this option, but need to be aware of this skewing.
- Tube-based acquisition from cytometers like the BD LSR-II is specifically designed to use polystyrene tubes (PS). This is because sample is forced up the sip tube by sealing the tube against a gasket, then pressuring the inside of the tube. Polystyrene plastic is rigid, and so pressurizes without deforming. Polystyrene is problematic, however, for any process that cultures immune cells in contact with it. Monocytes (MO) react to polystyrene by trying to phagocytose it, effectively flattening and adhering to the plastic. This complicates full recovery from any PS tubes used to culture suspensions with MOs. Since MO can be involved in antigen presentation in this assay, the tube plastic was switched to polypropylene, to which MO cannot adhere. This results in a technical complication, in that PP tubes do not have exactly the same dimension as PS tubes, and therefore do not form an airtight seal against the BD LSR-II sip assembly gasket. Using PS tubes with these cytometer therefore requires either transferring the finalized sample from PP to PS tubes, or else refitting the cytometer with a modified-dimension gasket (see Note 45).
- As a QA feature, when dealing with dozens, hundreds, or even thousands of tubes, we use tube racks equipped with a 'gripper' feature, which can hold each tube up in its slot. When we begin, the tubes are all pulled up. As we add reagent to a tube, we push it down. This way, we keep track of whether a given tube has been treated in a given step.
- Negative controls: If your antigens are all in DMSO, you might choose to put an equivalent amount of DMSO into your Negative control tube(s). This lab often has a diversity of test preparations, and so typically does not add solvent to its Negative control.
- Staphylococcus Enterotoxin B is a 'superantigen', capable of inducing a non-specific activation a large (e.g. 25%) fraction of T cells, accompanied by massive cytokine release from these cells. This is an effective positive control for this assay, because a failure to elicit massive TNFa or IFNg release strongly implies a technical mistake in execution. Because of its inherent danger, SEB is categorized as a Select Agent, and requires special paperwork to obtain (and then in only small quantities, ≤5mg). It also must be handled carefully. (Since SEB is possibly difficult to obtain, and requires special handling, a user can opt for any other antigen that researcher might know which is likely to reliably give a positive memory-recall response from the researchers cells. SEB is merely convenient and reliable, if available.)
- Because this assay does not have a reliable or consistent population of antigen-presenting cells, it is necessary to fully engage the lymphocyte receptors that would otherwise be triggered by the APCs (Antigen Presenting Cells, like macrophages, et al.). In the early work developing this assay, Picker, et al. evaluated the utility of a slate of antibodies against these costimulatory targets. They found that no single antibody target reliably yielded maximum effect, but that the combination of both anti-CD28 and anti-CD49d did.
- If a user of this assay wants to incorporate CD28 into the staining panel, a conjugated form of the antibody can be used, with one major caveat: Commercial off-the-shelf conjugated antibody reagents contain an amount of azide preservative that can be toxic to the cells in this assay. To get around this, we contracted for a custom high concentration (2 mg/ml) conjugate in low (0.05%) azide. (We obtained our custom stock from Beckman Coulter. More recently, we are revisiting the issue of whether the amount of azide in off-the-shelf reagents affects ICS results. Preliminary results suggest that amount of azide may not matter, but this testing is incomplete. Please email this author for future updates about this issue.)
- There is some controversy about the necessity of staining for the activation marker CD69. Some prominent groups have opted to omit this stain, and instead use the channel for another cytokine, or other marker. In our experience, however, this can result in data artifacts due to cytokine production from unactivated cells. Given the choice to add another cytokine, or ensure the cytokine data is reliable, we've opted for data reliability.
- Anti-CD45 is a very useful antibody to include in cases of 'dirty' samples. Example1: In lung wash, lymphocytes are a minority (1-6%) population, and mucous-trapped debris and non-WBC epithelial cells predominate. Example 2: In thawed cryopreserved PBMC, a large fraction of acquired events can be subcellular debris released from fractured cells. Example 3: In enzymatically-digested tissues like gut or liver, lymphocytes may be a minority population, and the process may have generated a lot of debris. In all these cases, an early gate of CD3 vs CD45 quickly removes the debris that would otherwise obscur downstream gating. In our experience, CD45 is required when doing plate-based ICS, because in the shorter fluid column of a plate well, all material (cells and debris) centrifuge to the bottom in a 2-3 min spin. In 5 ml tubes, in contrast, a 10 min spin does not reliably move all the small debris into the pellet. Over the multiple spins of this assay, tube-based ICS becomes largely clarified of small debris, whereas plate-based ICS never does.
- Staining for CD107ab offers a way to monitor the degranulation of CTL cells (cytotoxic lymphocyte). Use of this antibody necessitates swapping monensin for Brefeldin A. In our experience, this swap results in a relative reduction in cytokine signals.
- DMSO solutions melt slowly at room temperature, and so this reagent needs to be taken out to thaw at least 15 min before use. Moreover, DMSO is somewhat viscous, and so measurements of 1 μl are hard to do accurately. For this reason, and because of the advantages of using a repeater for delivery, we usually prepare dilutions of BfA in our tissue culture medium (49 μl R10: 1 μl BfA), and deliver 50 μl/tube. This volume will form a heavy enough drop on the side of a tube to reliably travel to the bottom of the tube. It is therefore possible to use a repeater, even touching the top of a tube, without great risk of cross-contamination. Such use of a repeater makes it possible to scale up the tube-based method.
- FACS Lysing Solution is a reagent invented by BD to do three things simultaneously: (1) Decontaminate a sample (e.g. HIV), (2) rupture and remove RBC (whose multi-sized aggregates can obscure and confound scatterplots, and (3) kill and fix the WBC of interest. FACS Lysing Solution does this by combining a hypotonic (weak) saline with paraformaldehyde (the small, fast-penetrating monomer of formaldehyde). The BD innovation (that continues making them money) was to titer these two components together, such that RBC swell and burst before they fix, whereas WBC fix before they burst. It is important to be be reliably consistent in the timing of this reagent application, because all unpermeabilized, intact cells in a hypotonic solution will continue to swell, thus the scatterplot size will change, if the Lyse is left on longer than 10 min. Perm does not have this problem, because porous cells will not swell due to osmotic pressure.
- Many investigators use the one-step reagent, BD CytoFix/CytoPerm, instead of the two-step Lyse, then Perm, so several comments about this are warranted. (1) CytoFix/CytoPerm is an appropriate reagent when dealing with samples lacking a significant presence of RBC (e.g. lung lavage, 'clean' PBMC, lymphocyte harvests from lymph node or gut biopsy). In our work the monkeys are often very sick due to complications of SIV infection, and hemolysis can result in very 'bloody' PBMC harvests. BAL samples can also sometimes be bloody. Given that our research almost always includes blood, our processing necessarily always requires RBC clearance, so we must use 'Lyse'. We therefore consistently apply it to all suspensions, regardless of the strict need. (2) Nonetheless, when we have compared the CFC results obtained from RBC-lacking samples processed with Lyse, then Perm, versus CytoFix/CytoPerm, we found that the former consistently gave larger signals than the latter (though the difference was small, typically a margin of approximately 10%). We attribute this better outcome to the irreversible Tween-20 being a superior permeant than the reversible saponon. (3) When we developed our plate-based CFC method, we realized that the well volumes were insufficient to allow RBC lysis within the wells. We therefore used ACD to remove RBCs prior to adding the cells to the plate. To counter that additional step, and to simplify the process, we substituted CytoFix/CytoPerm for the now-unnecessary 'Lyse'.
- The Picker Lab has worked for years with Tritech Research, a maker of non-water-jacketed CO2 incubators, in the development of a 37 °C, CO2 incubator that can rapidly cool to refrigerator temperature at specified clock times, in a way controlled and recorded by a computer. This was prompted by the frequent need, imposed by this CFC protocol, to transfer samples from a conventional incubator to a refrigerator, in the middle of the night (e.g. 3 am). Tritech is now selling a third generation of a single-chamber unit sized for tube racks (called the "UniBator"), and the third generation of a unit with six independently-controllable chambers for plate-based CFC (called the "HexaBator"). Although many incubators are available that can change temperature over a time frame of hours, as far as I know these are the only units capable of dropping from 37 °C to 4 °C in approximately 20 min (as required for consistent CFC results). As of this writing, Tritech has supplied units to the Picker Lab [3 UniBators, 3 HexaBators (all generations)], IAVI (1 UniBator, Gen1), VGTI-Florida/Watkins Lab (1 UniBator, Gen3), and VGTI-Oregon/Sacha Lab (1 UniBator, Gen3).
- Many researchers routinely use thawed cells for the CFC assay. Many use 'rest' periods after thawing, but before assay setup, to allow cells triggered for apoptosis to complete this process. Since we do nearly all our CFC with fresh cells, a reliable description of a post-thaw 'rest' and viability assessement can be found at Reference 2.
- As a general practice, all the reagents we use in the setup of this assay are equilibrated to room temperature before exposed to cells. A key parameter in our use of this assay is activation, and significant temperature changes to the cells (post-thaw) can affect their degree of activation. Also, the quality of this assay can be degraded by the use of tissue culture medium degraded in L-glutamine, or buffer-exhausted. So as a general practice, we use fresh-made medium for our setups.
- Adjustable pipettors are most-accurate in the middle of their range. When measuring 50 μl, for example, a P100 is a better choice than a P200 or P50. Also, DMSO solutions (e.g. peptide preparations) are more viscous than acqueous solutions, and so samples should be drawn and dispensed more slowly than acqueous solutions. Lastly, DMSO solutions have a tendency to form droplets on the outside of pipet tips, so after drawing in a measured amount of reagent, the tip should be dragged up the side of the vessel, to leave behind outside-adhering droplets. Failure to measure antigen accurately can result in large variations in outcomes.
- Angling the rack so that the tubes are nearly horizontal is crucial. At this orientation, the depth of the culture is shallower, facilitating better gas exchange. Also, the cells settle out over a much broader surface that would be the case if the tubes were upright. This appears to be a reason why tubes demonstrate better low-end sensitivity than plates (though this has not been experimentally proven). One hypothesis is that the looser, more-dispersed pellet that forms along the side of a tube is more optimal for CFC than is a more-compact pellet, as might form at the bottom of a U- or V-bottomed plate well.
- The 1 h pre-BfA interval is only necessary for non-peptide antigens, like proteins or lysates. This interval allows time for some processing to occur before adding the cytoskeleton-disrupting BfA. In unpublished examination, disrupting the pellet by vortexing after adding the BfA had a small, but minor negative effect on the size of antigen responses.
- In extensive but unpublished work, the Picker Lab examined the kinetics of antigen-stimulated cytokine secretion. We found that for TNFa, IFNg, and MIP1b, a sigmoidal accumulation of BfA-trapped cytokine signal plateaued from approximately 8-12 h. Beyond 12 h, signal decreased and non-Ag background noise increased. On the basis of this work, we've chosen to always run our CFC assays for 9 h total (1 h prior to BfA, 8 additional h with BfA). This timing yielded the most-consistent large signals for TNFa, IFNg, and MIP1b. In those same kinetic experiments, we found that IL2 signal emerges more quickly, maximizing from 4-6 h after setup, and degrading quickly after 6 h. For this reason, when we do CFC for which the IL2 signal is a very important parameter, we opt to do a 6 h total incubation, instead of a 9 h total incubation.
- This write-up describes four distinct ways of removing supernatants from tubes, because this is one of the dominant hand-manipulations involved in this workflow, and different methods are appropriate for different scale-ups. How one removes the supernatant, and how well, is vitally important to the quality of the produced data. The factors deserving attention include: (1) How much residual fluid is left behind? (2) Does the method disrupt and lose any of the cell pellet? (3) Does the method 'clean' the sample of sub-cellular debris? (4) Is the method fast? (5) Does the method require a lot of repetitive hand manipulation?
- We use a two-flask vacuum aspirator setup similar to that depicted below:
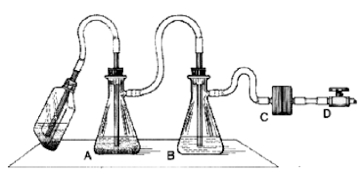
Figure 4. Aspirator trap assembly for use in a biosafety cabinet
In this setup, the aspirator wand goes to a primary trap, which has an overflow tube going to a secondary trap. The vacuum line connecting the secondary trap to the vacuum source has an in-line filter that stops air passage if it gets wet. This way, the vacuum pump never sucks sample material. The important reality to appreciate is that an aspiration wand causes significant air turbulence near its tip, and so to avoid mixing fluids near the pellet, the aspiration process must occur smoothly and fast. The operator should not 'dwell' near the pellet, or else the pellet will mix into the supernatant, and eventually be sucked up. One way to minimize this mixing is to affix a micropipettor tube (without aerosol plug) to the end of an aspiration wand. This significantly reduces air turbulence near the pellet, but it also significantly slows aspiration speed. A fast, effective hybrid approach is to combine use of a 'stopped' wand (see next comment), with a secondary 'clean-up' with a wand affixed with a pipettor tip.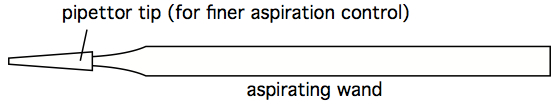
Figure 5. Aspiration wand, with pipettor tip for finer suction control - An 'stopped' aspirating wand is easily constructed by finding some Tygon tubing with an internal diameter close to the outer diameter of an aspirating wand. A short segment of this tubing can be split, so as to easily move it along the length of the wand. Trial and error can quckly identify the stop at which a smoothly-used wand leaves just the right amount of residual fluid, without any turbulence-stirring of the cell pellet. Once that stop location is determined, the split Tygon tubing stop can be taped shut, and prevented from moving with additional tape (see figure below). A stopped wand removes supernatant very quickly, and the tubes can be left in the centrifuge bucket insert, or rack, during this process. It still does one tube at a time, however, which may still be too slow and labor-intensive when dealing with hundreds of tubes simultaneously, however. Which is an argument for decanting (see next comment).
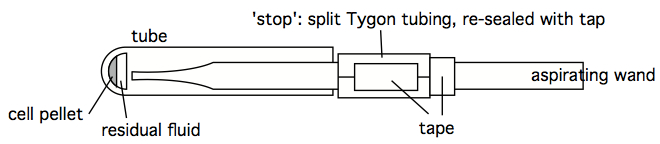
Figure 6. Stop on an aspirator wand, to enable rapid, 'no-look' aspiration when handling dozens or hundreds of tubes (note that this figure is better viewed at a 90o angle). - Decanting is just as it sounds, but is an art in practice. The keys to directly pouring out supernatant lie in (1) ensuring the centrifuge spin was long enough to set a tight, affixed pellet, (2) the subsequent processing doesn't 'bump' or vibrate the waiting tubes, loosening them from the tube bottom, (3) the pouring is smooth and fast and consistent in time and motion (leaving little time for the topmost material in the pellet to flow down and drain out of the tube), and (4) just the right amount of 'tap' to the drained tube to disengage the drop left at the end of the tube. Once mastered, the great benefit of decanting is that multiple tubes can be decanted simultaneously, and much faster than other methods. The potential pitfalls are sample loss and splashing (a big concern when dealing with biohazardous materials).
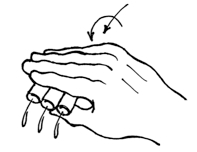
Figure 7. Hand-decanting supernatant from centrifuged ICS tubes - It is possible to scale this up even more radically by immobilizing all the tubes into a bucket insert using craft foam, as depicted in the figure below. Craft foam (available from arts-and-craft stores) can be easily cut to fit a bucket insert. Our inserts come as stackable layers, and we put foam between two of these layers. Where a tube would go, we first cut an 'X' bigger than the tube, then use a leather punch to remove a hole slightly smaller than a tube. This means that you can easily insert a tube, but that once inserted, the foam holds the tube in the slot. This way, an entire bucket of tubes can be dumped simultaneously. Afterwards, the entire bucket can be vortexed as a unit, but the process is even more an art than decanting handfuls of tubes. Crucially, it is important to know that the tubes at the edges of the insert vortex more effectively than those in the middle, so the insert needs to be vortexed in multiple,varying orientation for the process to be effective in all pellets.
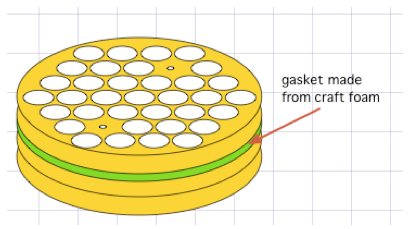
Figure 8. One example of how ICS tubes can be immobilized in a centrifuge bucket insert, so that decanting and vortexing can be consistently applied to an entire bucket of tubes simultaneously - Resuspending the pellets effectively, especially just prior to adding fixative (Lyse, Perm), is crucial to data quality. As with supernatant removal, we use several different methods, each appropriate to a particular scale of tube handling. For relatively few tubes, we hold several tubes in one hand, and flick/drag them over the fingers of the opposite hand several times (e.g. three times in one orientation, then rotate 90 degrees and draw them over again another three times). This is a very effective 'gold standard'. Alternatively, it is possible to drag the bottoms of the tubes against the front grate of a biosafety cabinet, rotating the tubes after half the drags, and this can achieve the same effect. An operator can also vortex individual tubes against a vortexer cup, or handfuls of tubes against a vortexer plate, but it is important to remember that if the tubes are held upright, the vortex created has its 'eye-of-the-hurricane' situated over the pellet one wants to disrupt. Therefore, one should always vortex the tubes at a (changing) angle, ideally after an initial flick to dislodge the pellet from the floor of the tube. Scaling up further, one can vortex an entire rack, or a gasketed bucket insert (like that depicted above), but this involves 'art', and one should practice it with pelleted RBCs to ensure one knows just how much vortexing, and how much orientation-changing is needed to ensure the pellets are fully and reliably disrupted.
- We long ago confirmed that cocktails of antibodies work as well as individually-administered single doses of antibody. Cocktails have the advantages of less hand-work, and more accurate measurements (large volumes are better-measured than small volumes). Moreover because staining kinetics is dependent on antibody concentration, we realized that consistent-volume cocktails give consistent results. For this reason, we typically formulate our cocktails such that we dispense 50 μl per tube. 50 μl reliably runs down the side of a PP tube, and is a volume effectively dispensed by a repeater.
- Initially, we always did staining on ice, in the dark. As we scaled up, this was one of the 'nice-ities'that was abandoned. When I investigated whether this has any negative effects on our quantitative results, I found none. Moreover, the staining is mostly done within 20 min, so a 30 min incubation is not sacred. Thirty min produces reliably-consistent good results, works well with large-scale processing, and allows for reasonable breaks (if needed).
- The Lyse step is the one that most-determines staining quality. If the cell pellet is ineffectively-disrupted, the paraformaldehyde will chemically fix clumped cells &/or cells and debris together. It also crucially affects the morphology of the lymphocytes, and hence the appearance of the key scatterplot display. The perm step tends not to be so pivotal, because fixed cells seem not to clump as avidly as live cells.
- This is a potential stopping point, if needed. At this point, the cells are fixed, and the paraformaldehyde is diluted, and so the tubes can sit days (e.g. a weekend) at 4 °C without any significant loss in quality. If we develop cytometer problems that will delay acquisition, we stop our stains at this point, so that at least the intracellular stains are as 'fresh' as possible before acquisition.
- It is not essential to disrupt the cell pellets afterwards. The goal is primarily to dilute the residual paraformaldehyde and Tween-20, and the volume held in the interstitial space is negligible to this process.
- It is important to do two washes, because the overall goal is to reduce-to-innocuous the amount of protein-fixing paraformaldehyde before adding the intracellular stain antibodies (which are proteins).
- Ki67 is a protein that transiently appears in the nucleus during mitosis. The staining pattern is a smear (rather than distinct negative and positive populations), and so obtaining useful timepoint comparisons is hugely dependent on the staining being absolutely consistent from timepoint to timepoint. To get this stain optimal, and consistent, it is necessary to use the smallest, brightest fluorophore (FITC, which penetrates to the nucleus best), and enough staining time (45 min) so that the staining process is reliably complete. Lastly, it is important that the samples be washed afterwards, with enough time in wash fluid so that anti-Ki67 MAb that has not bound to high-affinity sites has enough time to drift out of the cell (by the way, this is a disadvantage of CytoFix/CytoPerm, which requires the continuous presence of the permeabilizer,saponin, for intracellular staining. Once saponin is removed, whatever is inside the membrane is trapped. When Tween-20 is used, the membranes are dissolved away, such that low-affinity binding is lost during sample storage time, prior to acquisition. So sample quality can improve slightly over a day of storage).
- In the past,we added 1% paraformaldehyde to the tubes, to fix the intracellular antibodies in place. We came to realize this was not necessary (the high-affinity clones we were using had no noticeable off-rate in the < 3 days storage time), and that the addition of any fluid just increased our cytometer acquisition times.
- The exact purpose of beta-Mercaptoethanol is a bit unclear, and I've seen several different explanations. One is that it is a reducing agent that fights free radical oxygen species affecting lymphocytes. Another is that bME enhances the cysteine supply to the lymphoid cells, thereby increasing the intracellular level of tripeptide glutathione (Meister and Anderson, 1983). All seem to agree, however that empirically, bME improves tissue culture studies of lymphocytes (at least murine lymphocytes).
- CD45RA identifies naïve T cells, and is a stain recommended by the NHP ICS standards group, in their report of a plate-based method (Donaldson et al., 2012).
- CCR7 is found on T cells that require a secondary stimulus prior to displaying effector function. As with CD45RA, this is a stain recommended by the NHP ICS standards group (Donaldson et al., 2012).
- The Invitrogen Aqua LIVE/DEAD reagent stains protein, and distinguishes live and dead cells on the basis of how much protein the dye can access. Cells have approximately 1/100 as much protein on their surface as inside. A live cell has an intact membrane, and therefore takes up far less stain than a porous dead cell, which typically stains 2 logs brighter than a live cell.
- Cell phenotype markers like CD3, CD4, and CD8 can be stained either during the surface stain, or during the intracellar stain (CD45 MAb does not work on PFA-fixed antigen, so must be applied as a surface stain). For CD3 and CD8, there are both benefits and consequences staining these markers at the intracellular step. On the positive side, both receptors undergo internalization during antigen engagement, so responding cells become 'dimmer' in a surface stain. An inexperienced analyst could gate these cells out, unless s/he took into account the reduced surface brightness of responding cells. Applying these stains intracellularly obviates this problem, since the internalized receptors remain, and are available for staining. The drawback to this approach, however, is that all intracellular stains reduced the intensity separation between negative and positive populations.
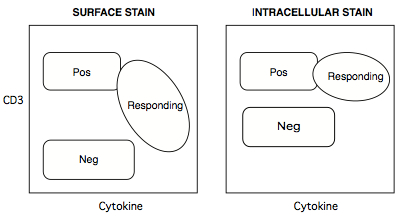
Figure 9. How intracellular staining of CD3+ T cells can solve the problem of CD3 internalization upon activation
Positive populations tend to stay put, but the negative population tends to be brighter for intracellular stains, because more low-affinity binding sites are available to antibodies accessing the insides of cells. - Most flow analysers push sample suspension up into their sip tube by pressurizing the sample tube. Because polystyrene is rigid, many analysers were designed for polystyrene tubes. On such cytometers (e.g. BD LSR-II), the tubes are fitted onto a sip assembly, with the tube wall forming an airtight seal against a gasket.
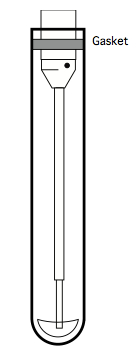
Figure 10. Illustration of the sip assembly of a BD LSR-II cytometric analyser, showing the gasket at the top, which is designed to seal against polystyrene tubes
Unfortunately, since monocytes and macrophages adhere themselves to PS, it is not appropriate for the CFC assay, since after a culture period any adherent cells will be unavailable for analysis. Consequently, a non-'sticky' plastic like polypropylene (PP) should be used (see Note 7). Standard PP tubes have a slightly different diameter than PS tubes, however, so the standard gasket on the sip assembly does not form an airtight seal against PP tubes. One easy solution to this problem is to widen a standard gasket by adding the outer ring of a second gasket, as illustrated below: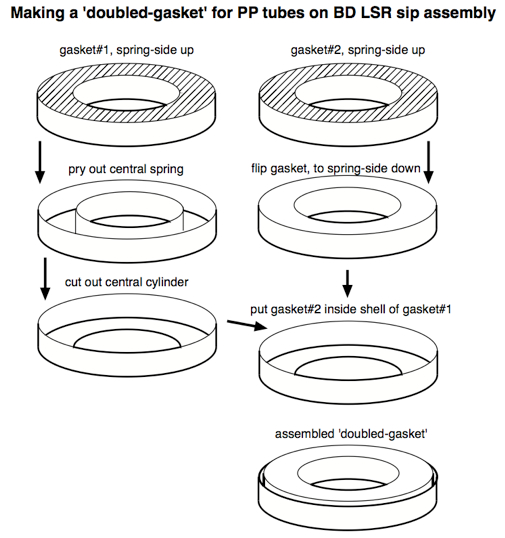
Figure 11. How an LSR-II sip assembly gasket can be modified to seal against polypropylene tubes (the type of plastic best for ICS) - In our experience, conventional CFC samples are fairly robust at 4 °C storage. Since the MAb-identified populations are mostly separated and discrete, the 'defocussing' that occurs during storage takes a while to affect gating outcomes. Our rule is that these samples should be acquired within 72 h, though use of some less-stable fluorophores might shorten this time. The inclusion of 'smeared' populations (e.g. Ki67, CD95) can also make it necessary to acquire sample data sooner.
- Because the possibility always exists that the cytometer will be unavailable (out-of-service, or fully-occupied) when samples need to be collected, we have investigated whether full-stained samples can be frozen, then thawed after a cytometer becomes available again. We have been surprised that this can sometimes work, though the scatterplot changes slightly. In our experience, most fluorescent results show no qualitative or quantitative changes. It is crucial to test this out before relying on this desperation move, however, because we have found that some fluorescent stains are, in fact, significantly changed. If one wanted this as a reliable fallback, various panels should be tested beforehand, and only ones robust through a freeze-thaw cycle should be used.
QA/QC Notes
'QA' stands for Quality Assurance, and 'QC' stands for Quality Control. QA refers to how you set a process up so as to maximize consistent quality, and minimize errors (or the consequence of mistakes). QC refers to post-process checking of the process outcome, to identify quality errors, and alert you that QA needs to be improved.
Some QA/QC notes relevant to the CFC assay:
- Training
- Prepare SOPs (Standard Operating Procedures).
- Have competent people train those who are learning.
- Proficiency test those who have been trained.
- Compare the outcomes of those who are competent with those learning.
- Compare the outcomes, occasionally, of those deemed competent (to identify method 'drift' issues that affect quality or consistency).
- Change the SOP to reflect current best practices.
- Compare the outcomes of those who are competent with those learning.
- Prepare SOPs (Standard Operating Procedures).
- Reagents
- All antibody reagents should be re-titered against the kind of cells you'll be staining (e.g. blood, PBMC, BAL, etc.; using an N ≥ 4).
- All process buffers should be at room temperature, and NOT warmed from 4 °C with a waterbath set at 37 °C. The reason for using buffers all at room temperature is (1) to avoid temperature shocks that induce activation, and (2) to keep the mammalian cells below the temperature at which they're metabolically active. Room temperature reliably does both.
- All antibody reagents should be re-titered against the kind of cells you'll be staining (e.g. blood, PBMC, BAL, etc.; using an N ≥ 4).
- Cells
- Freshly-harvested cells give more consistent results
- Thawed cells are variable for many reasons
- Variable effectiveness at cryopreservation
- Variable storage time, quality
- DMSO toxicity
- Variable thawing quality
Keep cells from warming to metabolic activity when in the presence of DMSO - Broken cell debris generated by the freeze-thaw (F/T) cycle, and adhering to live cells
- Intact carcasses of F/T-killed cells seeming like live cells (and artificially increasing the 'non-responding' denominator population
- Naïve cells are preferentially (but variably) lost during the F/T cycle
- Thawed cells are variable for many reasons
- The CFC assay is designed for 1e6 Ly/tube, but works as well for 0.5-2e6 Ly/tube, in 1 ml culture. If you reduce the culture volume, medium exhaustion and waste buildup in active cultures can variably affect result quality intra-run.
- Freshly-harvested cells give more consistent results
- Sample ID, labeling
- Always arrange samples in a logical, reproducible, consistently-applied order
- e.g. Our monkey IDs are 5-digit numbers
A random order: 25113, 25131, 23115, 25311
Numerically-ordered: 23115, 25113, 25131, 25311
This way, you can always ensure they're in a logical, reproducible order
- e.g. Our monkey IDs are 5-digit numbers
- Give samples two names, one that's a simplified shorthand easier for users to use
- e.g. A = 23115, B = 25113, C = 25131, D = 25311
- Or, if they're all in 'Group A': A1 = 23115, A2 = 25113, A3 = 25131, A4 = 25331
- This way, operators can deal with (A,B,C,D) or (A1,A2,A3,A4) instead of those confusing 5-digit numbers (which are just begging to get mixed-up).
- Also, if one number in an array is mis-transcribed, it is possible to deduce the error and correct it.
- e.g. A = 23115, B = 25113, C = 25131, D = 25311
- Number-identify antigens in a similar way
- e.g. 1 = Neg, 2 = X, 3 = Y, …7 = SEB
- In this way, if you orient the tubes in rack by this order, the first tube you handle will always be the negative control, and the last one will always be the antigen most likely to cause cross-contamination problems.
- This dual-identification,as noted above, helps definitively correct transcription errors.
- And this dual-identification has benefits in FlowJo and Excel.
FlowJo recognizes only the first 32 characters of a filename. If the 32nd character is 2=, you have a unique identification of that sample in a way you wouldn't if you were instead listing several SIV_( ) antigens.
After you export data from FlowJo, you end up with a filename which is a long text string, in which multiple discrete pieces of information are embedded. The Excel tool 'Text-To-Data' is useful for 'exploding' filenames into component parts, and characters like '=' are handy for this process.
- e.g. 1 = Neg, 2 = X, 3 = Y, …7 = SEB
- Use computer-printed labels
- Use a serif font (so that lower-case 'L' isn't confused with '1', for example).
- Use a font size large enough to be easily read.
- Use understood abbreviations, to allow for larger font sizes.
- Use label color to communicate grouping, timepoint, monkey, antigen, etc.
- Use a serif font (so that lower-case 'L' isn't confused with '1', for example).
- Always arrange samples in a logical, reproducible, consistently-applied order
- Process
- Use tube-gripping racks
'Tube UP' means step (e.g. cocktail addition) has not yet happened.
'Tube DOWN' means step completed. - Move reagent tubes laterally, or back/forward in their rack, to indicate whether they've been used yet.
- Prepare a checklist of steps, or reagents,and mark them off as the reagents are used, or steps completed.
- Use label placement on tubes to consistently orient them in racks.
- Reagent 1 can be dispensed down one wall.
- Reagent 2, during a following step, can be repeater-dispensed down a different wall, with reduced likelihood of cross-contamination.
- Reagent 1 can be dispensed down one wall.
- Organize samples so that the negative control tubes are always processed first, and the most-potent-antigen tubes are always processed last. This minimizes the consequences of any cross-contamination that does happen.
- Always process tubes in the same order. That increases the likelihood that they'll all get similar duration for each process (e.g. Lyse treatment, antibody exposure, etc).
- Make reagent cocktails, rather than delivering individual doses. Larger measurements are more accurate, and the fewer the measurements you make, the less likely you'll make a mistake.
- Habitually drag micropipettor tips up the side of a tube when you pull it out of a reagent vial. That way, you drag off any fluid holding on to the outside of the pipettor tip. This is especially important when measuring very small volumes of peptide antigen.
- Pipettors are most-accurate in their midrange. Use a P100 to measure 50 μl, rather than a P200 or P50.
- If possible, set up multiple negative controls, and use an average value for any background-subtraction.
- If confronted with so many samples that the processing cycle is too long to maintain quality, break the work up into parts that can be done with high quality. Sometimes, this issue applies to only a single step. For example, the CFC step most likely to cause quality problems is the addition of Lyse. If necessary, do sub-batches at a time, so as to maintain quality.
- Use tube-gripping racks
- Stain
- Use validated staining panels.
- Put the brightest fluorophores on the rarest events.
- Put the least-effective fluorophores on the most-plentiful &/or most-intense events.
- Be aware of issues like the spectral bleed of PE-Cy7 into the PE channel, and so put PE-Cy7 on rarer, dimmer events.
- Staining volume matters, so the use of consistently-50 μl cocktail volumes makes results more consistent.
- Use validated staining panels.
- Acquisition
- Samples should be acquired within 72 h.
- In our experience samples stained with Alexa-700 suffer rapid quality erosion, and should be acquired within 24 h.
- If cytometer backlog or dysfunction mean some CFC samples will wait longer than 72 h before acquisition, the staining process can be stopped after Lyse,and the intracellular stain delayed until just before acquisition.
- Samples should be acquired within 72 h.
- Equipment
- Each piece of critical equipment should be assigned to a staff member, who is given responsibility to monitor, maintain, performance-assess, and repair.
- Coulter counters reliably count only blood samples, and are notoriously inaccurate for anything else. They can make hugely-variable, hugely-inaccurate counts with debris-rich samples like BAL, thawed cells, and enzymatically-digested solid tissues.
- Flow cytometers:
- Cytometers should be monitored by the BD CST system (or a functionally equivalent performance-monitoring system).
- Lasers should be warmed at least 45 minutes before use.
- Fluidics should get cleaned at least once a day (and again, if a lot of 'dirty' samples are run, leading to buildups that change the time delay).
- Compensation:
Use compensation beads, rather than stained cells (Beads are brighter.).
Make fresh compensation bead samples weekly.
Make compensation samples for all antibody reagents used (This is especially important for the tandem dyes, like PerCP-Cy5.5, PE-TexasRed, PE-Cy7, APC-Cy7, etc.).
Compensate each experiment anew (rather than using compensation matrices from weeks before). - Acquisition speed matters
Discrete populations can gateably-tolerate 'blurring' better than smears. Therefore CFC without CD28 v CD95, or Ki67 gating can be run faster with that gating.
- Cytometers should be monitored by the BD CST system (or a functionally equivalent performance-monitoring system).
- Each piece of critical equipment should be assigned to a staff member, who is given responsibility to monitor, maintain, performance-assess, and repair.
Recipes
- Aqua LIVE/DEAD kit
- Concentrated dye stock
1 vial powder dissolved in 50 μl DMSO, can stored as frozen aliquots for weeks - Staining Solution (made fresh)
- Dilute 2 μl conc dye into 78 μl deionized water (DI)
- Dilute 50 μl of the solution in step 5bi into 950 μl 1x PBS
- Stored on ice prior to use
- Dilute 2 μl conc dye into 78 μl deionized water (DI)
- Concentrated dye stock
- 'R10' tissue culture medium
- RPMI-1640 (1x) (w/o L-glutamine, 0.1 μm filtered)
- Fetal Bovine Serum (defined, heat-inactivated, 40 nm-filtered)
Stored as frozen aliquots - Penicillin+Streptomycin (P/S) Solution
Stored as frozen aliquots - L-glutamine (200 mM)
Stored as frozen aliquots; light-sensitive when thawed - Sodium pyruvate (SP)
- beta-Mercaptoethanol (bME) (Note 40)
Light sensitive; stored in foil-wrapped bottle - Sterile-filtration apparatus 500 ml capacity 0.22 μm cellulose-acetate filter
- Formulation
- Thaw frozen components
- Combine additives in top of filter system, then filter
50 ml FBS
10 ml P/S
10 ml L-glutamine
5 ml SP - Rinse filter with sterile RPMI
- Directly pour additional RPMI into the filtrate, to 500 ml
- While swirling the R10, slowly add 500 μl bME
- Cap tightly; protect from light; stored at 4 °C
- Thaw frozen components
- RPMI-1640 (1x) (w/o L-glutamine, 0.1 μm filtered)
- 'PAB' (phosphate albumin buffer) wash buffer
- DPBS, makes 10 L at 1x, or 1 L at 10x
- Bovine serum albumin (BSA)
- Sodium azide (preservative; NaN3)
- Highly toxic (Wear mask when measuring!)
- Deionized, filtered water (because of the azide, autoclaving not needed)
- Formulation of 10x stock:
- Dispense 4 L of DI water into a 10 L carbuoy
- Add a sterilized stirbar
- A top a magnetic stirrer, start a gentle swirl
- Add 50 g of BSA slowly to on the top of the swirl (avoid clumps)
- Add 22.75 g of NaN3, slowly
- Stir overnight, at room temperature
- Next day, after all previous additives are dissolved
- Add contents of five bottles of powdered DPBS (each bottle makes 1 L at 10x)
- Rinse bottles with fluid from the carbuoy, then return
- Increase stir speed; check every 2 h until all is dissolved
- Dispense 10x solution into sterile 1 L bottles; cap tightly
- Store bottles at 4 °C
- Dispense 4 L of DI water into a 10 L carbuoy
- Diluting to 1x working stock
- Into an emptied and sterilized 20 L carbuoy, pour two 1 L bottles of 10x PAB stock
- Rinse the emptied 1 L bottles with DI water, return
- Add DI water to carbuoy until the total volume is 20 L
- Cap, then mix by inversion
- Stored carbuoy at 4 °C
- Using 1x working stock
- Transfer to bottle with pump dispenser (e.g. Brinkmann Bottletop Dispenser; e.g., Brand Tech Dispensette)
- Use PAB at room 4 °C or room temperature
- Into an emptied and sterilized 20 L carbuoy, pour two 1 L bottles of 10x PAB stock
- DPBS, makes 10 L at 1x, or 1 L at 10x
- 'Lyse' fixation and RBC-lysing solution (Note 17)
- BD FACS Lysing Solution, 10x concentrate
Note: Contains paraformaldehyde. - Deionized water
- Formulation
- Combine ingredients in the following proportions
- 50 ml 10x FACS Lysing Solution
- 450 ml DI water
- Mix by shaking, stirring, or inversion
- Dispense into dark plastic bottles
- Store tightly capped bottles at room temperature
- QC test with whole blood for acceptable outcome
- Combine ingredients in the following proportions
- BD FACS Lysing Solution, 10x concentrate
- 'Perm' (1x) fixation and irreversible cell-permeating solution (Note 18)
- 1x 'Lyse' (from above)
- Tween-20 (polyoxyethylenesorbitan monolaurate)
- This requires a high-viscosity pipettor to measure well
- Formulation
- To 1 L of 'Lyse' add 500 μl Tween-20 while stirring
- Stir atop a magnetic stirrer overnight
- Dispense into dark plastic bottles
- Store tighly capped bottles at room temperature
- QC test with Ki67 antibody, for acceptable outcome
- To 1 L of 'Lyse' add 500 μl Tween-20 while stirring
- 1x 'Lyse' (from above)
Acknowledgments
The methods described in this protocol have been evolving since the 1995 paper cited in this protocol (Picker et al., 1995), and were used in the two 2013 papers published by our group (Hansen et al., 2013a and Hansen et al., 2013b). Funding has been provided by NIH and by the Bill and Melinda Gates Foundation.
References
- Andersson, U., Hallden, G., Persson, U., Hed, J., Moller, G. and DeLey, M. (1988). Enumeration of IFN-γ-producing cells by flow cytometry. Comparison with fluorescence microscopy. J Immunol Methods 112(1): 139-142.
- Donaldson, M. M., Kao, S. F., Eslamizar, L., Gee, C., Koopman, G., Lifton, M., Schmitz, J. E., Sylwester, A. W., Wilson, A., Hawkins, N., Self, S. G., Roederer, M. and Foulds, K. E. (2012). Optimization and qualification of an 8-color intracellular cytokine staining assay for quantifying T cell responses in rhesus macaques for pre-clinical vaccine studies. J Immunol Methods 386(1-2): 10-21.
- Foulds, K. E., Donaldson, M. and Roederer, M. (2012). OMIP-005: Quality and phenotype of antigen-responsive rhesus macaque T cells. Cytometry A 81(5): 360-361.
- Fukazawa, Y., Park, H., Cameron, M. J., Lefebvre, F., Lum, R., Coombes, N., Mahyari, E., Hagen, S. I., Bae, J. Y., Reyes, M. D., 3rd, Swanson, T., Legasse, A. W., Sylwester, A., Hansen, S. G., Smith, A. T., Stafova, P., Shoemaker, R., Li, Y., Oswald, K., Axthelm, M. K., McDermott, A., Ferrari, G., Montefiori, D. C., Edlefsen, P. T., Piatak, M., Jr., Lifson, J. D., Sekaly, R. P. and Picker, L. J. (2012). Lymph node T cell responses predict the efficacy of live attenuated SIV vaccines. Nat Med 18(11): 1673-1681.
- Gardner, M. B. (1989). SIV infected rhesus macaques: an AIDS model for immunoprevention and immunotherapy. Adv Exp Med Biol 251: 279-293.
- Hansen, S. G., Vieville, C., Whizin, N., Coyne-Johnson, L., Siess, D. C., Drummond, D. D., Legasse, A. W., Axthelm, M. K., Oswald, K., Trubey, C. M., Piatak, M., Jr., Lifson, J. D., Nelson, J. A., Jarvis, M. A. and Picker, L. J. (2009). Effector memory T cell responses are associated with protection of rhesus monkeys from mucosal simian immunodeficiency virus challenge. Nat Med 15(3): 293-299.
- Hansen, S. G., Ford, J. C., Lewis, M. S., Ventura, A. B., Hughes, C. M., Coyne-Johnson, L., Whizin, N., Oswald, K., Shoemaker, R., Swanson, T., Legasse, A. W., Chiuchiolo, M. J., Parks, C. L., Axthelm, M. K., Nelson, J. A., Jarvis, M. A., Piatak, M., Jr., Lifson, J. D. and Picker, L. J. (2011). Profound early control of highly pathogenic SIV by an effector memory T-cell vaccine. Nature 473(7348): 523-527.
- Hansen, S. G., Sacha, J. B., Hughes, C. M., Ford, J. C., Burwitz, B. J., Scholz, I., Gilbride, R. M., Lewis, M. S., Gilliam, A. N., Ventura, A. B., Malouli, D., Xu, G., Richards, R., Whizin, N., Reed, J. S., Hammond, K. B., Fischer, M., Turner, J. M., Legasse, A. W., Axthelm, M. K., Edlefsen, P. T., Nelson, J. A., Lifson, J. D., Fruh, K. and Picker, L. J. (2013). Cytomegalovirus vectors violate CD8+ T cell epitope recognition paradigms. Science 340(6135): 1237874.
- Hansen, S. G., Piatak, M., Jr., Ventura, A. B., Hughes, C. M., Gilbride, R. M., Ford, J. C., Oswald, K., Shoemaker, R., Li, Y., Lewis, M. S., Gilliam, A. N., Xu, G., Whizin, N., Burwitz, B. J., Planer, S. L., Turner, J. M., Legasse, A. W., Axthelm, M. K., Nelson, J. A., Fruh, K., Sacha, J. B., Estes, J. D., Keele, B. F., Edlefsen, P. T., Lifson, J. D. and Picker, L. J. (2013). Immune clearance of highly pathogenic SIV infection. Nature 502(7469): 100-104.
- Jung, T., Schauer, U., Heusser, C., Neumann, C. and Rieger, C. (1993). Detection of intracellular cytokines by flow cytometry. J Immunol Methods 159(1-2): 197-207.
- McClure, H. M., Anderson, D. C., Fultz, P. N., Ansari, A. A., Lockwood, E. and Brodie, A. (1989). Spectrum of disease in macaque monkeys chronically infected with SIV/SMM. Vet Immunol Immunopathol 21(1): 13-24.
- Picker, L. J., Singh, M. K., Zdraveski, Z., Treer, J. R., Waldrop, S. L., Bergstresser, P. R. and Maino, V. C. (1995). Direct demonstration of cytokine synthesis heterogeneity among human memory/effector T cells by flow cytometry. Blood 86(4): 1408-1419.
- Pitcher, C. J., Hagen, S. I., Walker, J. M., Lum, R., Mitchell, B. L., Maino, V. C., Axthelm, M. K. and Picker, L. J. (2002). Development and homeostasis of T cell memory in rhesus macaque. J Immunol 168(1): 29-43.
- Prussin, C. and Metcalfe, D. D. (1995). Detection of intracytoplasmic cytokine using flow cytometry and directly conjugated anti-cytokine antibodies. J Immunol Methods 188(1): 117-128.
- Sander, B., Andersson, J. and Andersson, U. (1991). Assessment of cytokines by immunofluorescence and the paraformaldehyde-saponin procedure. Immunol Rev 119: 65-93.
- Schuerwegh, A. J., Stevens, W. J., Bridts, C. H. and De Clerck, L. S. (2001). Evaluation of monensin and brefeldin A for flow cytometric determination of interleukin-1β, interleukin-6, and tumor necrosis factor-alpha in monocytes. Cytometry 46(3): 172-176.
- Waldrop, S. L., Pitcher, C. J., Peterson, D. M., Maino, V. C. and Picker, L. J. (1997). Determination of antigen-specific memory/effector CD4+ T cell frequencies by flow cytometry: evidence for a novel, antigen-specific homeostatic mechanism in HIV-associated immunodeficiency. J Clin Invest 99(7): 1739-1750.
Article Information
Copyright
© 2014 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Sylwester, A. W., Hansen, S. G. and Picker, L. J. (2014). Quantification of T Cell Antigen-specific Memory Responses in Rhesus Macaques, Using Cytokine Flow Cytometry (CFC, also Known as ICS and ICCS): from Assay Set-up to Data Acquisition. Bio-protocol 4(8): e1110. DOI: 10.21769/BioProtoc.1110.
Category
Immunology > Immune cell function > Antigen-specific response
Immunology > Immune cell staining > Flow cytometry
Cell Biology > Cell-based analysis > Flow cytometry
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.