

热门实验
过去三个月内浏览量最高的文章
Click-qPCR: A Simple Tool for Interactive qPCR Data Analysis
Click-qPCR:一种用于交互式 qPCR 数据分析的简便工具

Real-time quantitative PCR (qPCR) is a pivotal technique for analyzing gene expression and DNA copy number variations. However, the limited availability of user-friendly software tools for qPCR data analysis presents a significant challenge for experimental biologists with limited computational skills. To address this issue, we developed Click-qPCR, a user-friendly and web-based Shiny application for qPCR data analysis. Click-qPCR streamlines ΔCq and ΔΔCq calculations using user-uploaded CSV data files. The interactive interface of the application allows users to select genes and experimental groups and perform Welch’s t tests and one-way analysis of variance with Dunnett’s post-hoc test for pairwise and multi-group comparisons, respectively. Results are visualized via interactive bar plots (mean ± standard deviation with individual data points) and can be downloaded as publication-quality images, along with summary statistics. Click-qPCR empowers researchers to efficiently process, interpret, and visualize qPCR data regardless of their programming experience, thereby facilitating routine analysis tasks. Click-qPCR Shiny application is available at https://kubo-azu.shinyapps.io/Click-qPCR/, while its source code and user guide are available at https://github.com/kubo-azu/Click-qPCR.
Cycloheximide (CHX) Chase Assay to Examine Protein Half-life
环己酰亚胺(CHX)追踪测定法检测蛋白质半衰期
Cycloheximide (CHX) is a small molecule derived from Streptomyces griseus that acts as fungicide. As a ribosome inhibitor, CHX can restrict the translation elongation of eukaryotic protein synthesis. Once protein synthesis is inhibited by CHX, the level of intracellular proteins decreases by degradation through the proteasome or lysosome system. Thus, the CHX chase assay is widely recognized and used to observe intracellular protein degradation and to determine the half-life of a given protein in eukaryotes. Here, we present a complete experimental procedure of the CHX chase assay.Graphical overview

Room-Temperature Storage of Zebrafish and Medaka Sperm Using Lactic Acid-Stabilized L-15 Medium
利用乳酸稳定的 L-15 培养基实现斑马鱼和青鳉精子的室温保存
Zebrafish offer numerous advantages as a vertebrate model because of their rapid development, high fecundity, transparent embryos, and ease of genetic manipulation. A wide variety of transgenic and mutant fish lines have been generated, and efficiently sharing these resources is crucial for advancing research. Zebrafish lines have typically been exchanged as early embryos, adult fish, or cryopreserved sperm, making transportation costly and logistically challenging. Here, we provide a protocol for preserving functional zebrafish sperm for more than 7 days at room temperature and subsequent in vitro fertilization using the preserved sperm. In this protocol, sperm collected either from the cloaca of an anesthetized male or from dissected testes is stored in L-15-based storage medium. Importantly, the storage medium, originally developed for zebrafish, is also applicable to medaka, another widely used vertebrate model. This sperm storage method allows researchers to ship sperm using low-cost methods and to investigate key factors for motility and fertilizing ability in those sperm.
Generation of Human Induced Pluripotent Stem Cell (hiPSC)-Derived Astrocytes for Amyotrophic Lateral Sclerosis and Other Neurodegenerative Disease Studies
产生人类诱导多能干细胞 (hiPSC) 衍生的星形胶质细胞,用于研究肌萎缩侧索硬化症和其他神经退行性疾病
Astrocytes are increasingly recognized for their important role in neurodegenerative diseases like amyotrophic lateral sclerosis (ALS). In ALS, astrocytes shift from their primary function of providing neuronal homeostatic support towards a reactive and toxic role, which overall contributes to neuronal toxicity and cell death. Currently, our knowledge on these processes is incomplete, and time-efficient and reproducible model systems in a human context are therefore required to understand and therapeutically modulate the toxic astrocytic response for future treatment options. Here, we present an efficient and straightforward protocol to generate human induced pluripotent stem cell (hiPSC)-derived astrocytes implementing a differentiation scheme based on small molecules. Through an initial 25 days, hiPSCs are differentiated into astrocytes, which are matured for 4+ weeks. The hiPSC-derived astrocytes can be cryopreserved at every passage during differentiation and maturation. This provides convenient pauses in the protocol as well as cell banking opportunities, thereby limiting the need to continuously start from hiPSCs. The protocol has already proven valuable in ALS research but can be adapted to any desired research field where astrocytes are of interest.Key features• This protocol requires preexisting experience in hiPSC culturing for a successful outcome.• The protocol relies on a small molecule differentiation scheme and an easy-to-follow methodology, which can be paused at several time points.• The protocol generates >50 × 106 astrocytes per differentiation, which can be cryopreserved at every passage, ensuring a large-scale experimental output.Graphical overview

A Guide to Basic RNA Sequencing Data Processing and Transcriptomic Analysis
基础RNA测序数据处理与转录组分析指南
RNA sequencing (RNA-Seq) has transformed transcriptomic research, enabling researchers to perform large-scale inspection of mRNA levels in living cells. With the growing applicability of this technique to many scientific investigations, the analysis of next-generation sequencing (NGS) data becomes an important yet challenging task, especially for researchers without a bioinformatics background. This protocol offers a beginner-friendly step-by-step guide to analyze NGS data (starting from raw .fastq files), providing the required codes with an explanation of the different steps and software used. We outline a computational workflow that includes quality control, trimming of reads, read alignment to the genome, and gene quantification, ultimately enabling researchers to identify differentially expressed genes and gain insights on mRNA levels. Multiple approaches to visualize this data using statistical and graphical tools in R are also described, allowing the generation of heatmaps and volcano plots to represent genes and gene sets of interest.
A Simple, Reproducible Procedure for Chemiluminescent Western Blot Quantification
一种简单、可重复的化学发光蛋白质印迹定量方法
Western blotting is a universally used technique to identify specific proteins from a heterogeneous and complex mixture. However, there is no clear and common procedure to quantify the results obtained, resulting in variations due to the different software and protocols used in each laboratory. Here, we have developed a procedure based on the increase in chemiluminescent signal to obtain a representative value for each band to be quantified. Images were processed with ImageJ and subsequently compared using R software. The result is a linear regression model in which we use the slope of the signal increase within the combined linear range of detection to compare between samples. This approach allows to quantify and compare protein levels from different conditions in a simple and reproducible way. Graphical overview
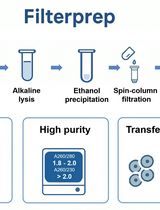
Plasmid DNA Purification Using Filterprep With an Optional Endotoxin Removal Step
基于 Filterprep 的质粒 DNA 纯化方法及可选内毒素去除步骤
This protocol presents a modified version of the Filterprep method originally reported in New Biotechnology, adding an optional step to reduce endotoxin levels. Filterprep is a simple, rapid, and cost-effective approach to plasmid DNA purification that couples ethanol precipitation with a single spin-column filtration step, eliminating chaotropic salts and silica binding. The formulations and parameters are fully transparent and do not rely on proprietary buffers, using only standard laboratory reagents and widely available miniprep columns. Under matched conditions, the method recovers high-purity plasmid DNA with yields up to fivefold higher than those obtained with representative commercial midiprep kits. The workflow is readily adoptable in most molecular biology laboratories and, under routine conditions, can be completed in approximately 40 min. The resulting DNA is suitable for molecular cloning, PCR, sequencing, and other downstream biochemical applications. Endotoxin is a lipopolysaccharide (LPS) found in the outer membrane of Gram-negative bacteria and may carry over during plasmid preparation. For experiments requiring lower endotoxin input, an optional modification resuspends the DNA pellet in a Triton X-114 wash buffer before column loading to decrease lipopolysaccharide carryover. The method is modular and extensible, allowing adjustment of precipitation and wash conditions, variation in the number of washes, selection of alternative column formats, and integration of endotoxin-reduction modules without altering the core principle. These features facilitate troubleshooting and quality control, enable scaling from routine batches to larger culture volumes and higher throughput, and allow seamless integration with existing workflows.
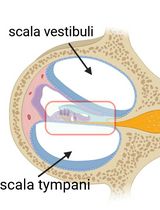
Cochlear Organ Dissection, Immunostaining, and Confocal Imaging in Mice
小鼠耳蜗器官解剖、免疫染色及共聚焦成像
The organ of Corti, located in the inner ear, is the primary organ responsible for animal hearing. Each hair cell has a V-shaped or U-shaped hair bundle composed of actin-filled stereocilia and a kinocilium supported by true transport microtubules. Damage to these structures due to noise exposure, drug toxicity, aging, or environmental factors can lead to hearing loss and other disorders. The challenge when examining auditory organs is their location within the bony labyrinth and their small and fragile nature. This protocol describes the dissection procedure for the cochlear organ, followed by confocal imaging of immunostained endogenous and fluorescent proteins. This approach can be used to understand hair cell physiology and the molecular mechanisms required for normal hearing.
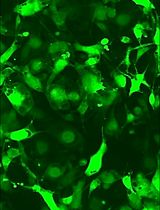
A Protocol to Induce Brown and Beige Adipocyte Differentiation From Murine and Human Adipose-Derived SVF
诱导小鼠与人源脂肪来源 SVF 分化为棕色与米色脂肪细胞的操作流程
Adipose cells vary functionally, with white adipocytes storing energy and brown/beige adipocytes generating heat. Mouse and human subcutaneous white adipose tissue (WAT)-derived stromal vascular fraction (SVF) provides mesenchymal stem cells (MSCs) that can be differentiated into thermogenic adipocytes using pharmacological cocktails. After six days of browning induction, these cells exhibited significant upregulation of thermogenic markers (UCP1, Cidea, Dio2, PRDM16) along with adipogenic genes (PPARγ, aP2), showing enhanced thermogenic potential. This in vitro system offers a practical platform to study adipogenesis and thermogenic regulation.
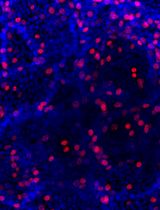
An Optimized Protocol for Simultaneous Propagation of Patient-derived Organoids and Matching CAFs
优化的患者来源类器官与匹配癌相关成纤维细胞同步培养方案
Recurrent hormone receptor-positive (HR+) breast cancer is a leading cause of cancer mortality in women. Recurrence and resistance to targeted therapies have been difficult to study due to the long clinical course of the disease, the complex nature of resistance, and the lack of clinically relevant model systems. Existing models are limited to a few HR+ cell lines, organoid models, and patient-derived xenograft models, all lacking components of the human tumor microenvironment. Furthermore, the low take rate and loss of estrogen receptor (ER) expression in patient-derived organoids (PDOs) has been challenging. Our protocol allows simultaneous isolation of PDOs and matching cancer-associated fibroblasts (CAFs) from primary and metastatic HR+ breast cancers. Importantly, our protocol has a higher take rate and enables long-term culturing of PDOs that retain ER expression. Our matching PDOs and CAFs will provide researchers with a new resource to study the influence of the tumor microenvironment on various aspects of cancer biology such as cell growth and drug resistance in HR+ breast cancer.
Preparation and Characterization of Lipid Nanoparticles Co-loaded With DNA and Nitro-Oleic Acid
DNA 与硝基油酸共载脂质纳米颗粒的制备与表征
Lipid nanoparticles (LNPs) are powerful carriers for nucleic acid delivery, but plasmid DNA-loaded LNPs (pDNA-LNPs) have been limited by inflammation and toxicity. We showed that standard pDNA-LNPs activate the cGAS–STING pathway, leading to severe immune responses and mortality in mice. To overcome this, we co-loaded nitro-oleic acid (NOA), an endogenous STING inhibitor, into pDNA-LNPs. NOA-pDNA-LNPs mitigated inflammation, enabled safe in vivo delivery, and supported sustained gene expression for months. Here, we present a detailed protocol for producing and characterizing NOA-pDNA-LNPs to facilitate safer, long-term gene delivery applications.
Cloning a Chloroplast Genome in Saccharomyces cerevisiae and Escherichia coli
在酿酒酵母和大肠杆菌中克隆叶绿体基因组
Chloroplast genomes present an alternative strategy for large-scale engineering of photosynthetic eukaryotes. Prior to our work, the chloroplast genomes of Chlamydomonas reinhardtii (204 kb) and Zea mays (140 kb) had been cloned using bacterial and yeast artificial chromosome (BAC/YAC) libraries, respectively. These methods lack design flexibility as they are reliant upon the random capture of genomic fragments during BAC/YAC library creation; additionally, both demonstrated a low efficiency (≤ 10%) for correct assembly of the genome in yeast. With this in mind, we sought to create a highly flexible and efficient approach for assembling the 117 kb chloroplast genome of Phaeodactylum tricornutum, a photosynthetic marine diatom. Our original article demonstrated a PCR-based approach for cloning the P. tricornutum chloroplast genome that had 90%–100% efficiency when screening as few as 10 yeast colonies following assembly. In this article, we will discuss this approach in greater depth as we believe this technique could be extrapolated to other species, particularly those with a similar chloroplast genome size and architecture.
In Vitro Bone Marrow–Derived Dendritic Cells (BMDC) Generation for Antigen Presentation Assay
体外诱导骨髓来源树突状细胞(BMDC)用于抗原呈递实验

Dendritic cells (DC) are sentinel cells of the immune system that process and present antigens to activate T cells, thus serving to bridge the innate and adaptive immune systems. DCs are particularly efficient at cross-presentation whereby exogenously acquired antigens are processed and presented in context with MHCI molecules to activate CD8+ T cells. Assaying antigen presentation by DCs is a critical parameter in assessing immune functionality. However, the low abundance of bona fide DCs within the lymphoid compartments limits the utility of such assays. An alternative approach employing the culturing of bone marrow cells in the presence of factors needed for DC lineage commitment can result in the differentiation of bone marrow dendritic cells (BMDCs). This protocol details the process of in vitro generation of BMDCs and demonstrates their subsequent utility in antigen presentation assays. The protocol described can be adapted to various conditions and antigens.

Egg Microinjection for the Silkworm Bombyx mori
家蚕卵的显微注射操作方法
The silkworm Bombyx mori has been extensively utilized in sericulture and serves as a representative model insect of Lepidoptera in various fields of life sciences and applied research. In recent years, its significance has further increased in molecular genetics and functional genomics. Germline transformation and genome editing in B. mori require the injection of vector solutions into early embryos; however, the thick eggshell of B. mori presents a significant challenge for microinjection. Conventional methods involve arranging eggs, pre-pierced with a tungsten needle, followed by solution injection, making the process both time-consuming and technically demanding. Here, we describe a simplified and more efficient microinjection protocol. Unlike conventional approaches, our method eliminates the need for egg removal from the egg-laying sheet and egg alignment on the slide glass by allowing injections to be performed directly on eggs retained on the egg-laying sheet. A thick-walled glass capillary, capable of penetrating the rigid eggshell, is used to directly pierce the eggshell and deliver the solution. By eliminating the need for egg alignment and micromanipulator operation, this protocol significantly enhances efficiency, enabling higher-throughput embryo injections within a shorter time frame. Moreover, this approach holds potential for application to other insect species with similarly thick eggshells.
Reproducible Emu-Based Workflow for High-Fidelity Soil and Plant Microbiome Profiling on HPC Clusters
基于 Emu 的可重复计算流程:在高性能计算集群上实现高保真土壤—植物微生物组解析
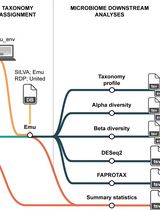
Accurate profiling of soil and root-associated bacterial communities is essential for understanding ecosystem functions and improving sustainable agricultural practices. Here, a comprehensive, modular workflow is presented for the analysis of full-length 16S rRNA gene amplicons generated with Oxford Nanopore long-read sequencing. The protocol integrates four standardized steps: (i) quality assessment and filtering of raw reads with NanoPlot and NanoFilt, (ii) removal of plant organelle contamination using a curated Viridiplantae Kraken2 database, (iii) species-level taxonomic assignment with Emu, and (iv) downstream ecological analyses, including rarefaction, diversity metrics, and functional inference. Leveraging high-performance computing resources, the workflow enables parallel processing of large datasets, rigorous contamination control, and reproducible execution across environments. The pipeline’s efficiency is demonstrated on full-length 16S rRNA gene datasets from yellow pea rhizosphere and root samples, with high post-filter read retention and high-resolution community profiles. Automated SLURM scripts and detailed documentation are provided in a public GitHub repository (https://github.com/henrimdias/emu-microbiome-HPC; release v1.0.2, emu-pipeline-revised) and archived on Zenodo (DOI: 10.5281/zenodo.17764933).
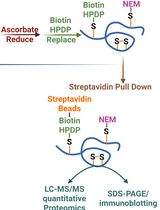
Quantitative Proteomics of Nitrosylated Proteins in Melanoma Using the Biotin-Switch Technique Combined With Tandem Mass Tag Labeling
结合生物素开关技术与 TMT 标记的黑色素瘤蛋白硝基化定量蛋白组学分析
Protein S-nitrosylation is a critical post-translational modification that regulates diverse cellular functions and signaling pathways. Although various biochemical methods have been developed to detect S-nitrosylated proteins, many suffer from limited specificity and sensitivity. Here, we describe a robust protocol that combines a modified biotin-switch technique (BST) with streptavidin-based affinity enrichment and quantitative mass spectrometry to detect and profile nitrosylated proteins in cultured cells. The method involves blocking free thiols, selective reduction of nitrosothiols, biotin labeling, enrichment of biotinylated proteins, and identification by tandem mass tag (TMT)-based quantitative mass spectrometry. Additionally, site-directed mutagenesis is employed to generate “non-nitrosylable” mutants for functional validation of specific nitrosylation sites. This protocol provides high specificity, quantitative capability, and versatility for both targeted and global analysis of protein nitrosylation.
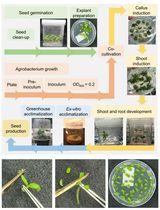
Highly Efficient Agrobacterium-Mediated Transformation of Tomato cv Micro-Tom From Cotyledon Explants
基于子叶外植体的 Micro-Tom 番茄高效根癌农杆菌转化方法
The tomato (Solanum lycopersicum) is a widely cultivated crop worldwide that serves as a model system for fruit development studies. Agrobacterium tumefaciens–mediated transformation of tomato has played a central role as a tool for analyzing the function of candidate genes and producing transgenic lines with enhanced resistance to pathogens, tolerance to abiotic stresses, and improved fruit quality traits. Among the many tomato varieties, the miniature dwarf cultivar Micro-Tom (MT) has been increasingly adopted as a model system for tomato research due to its short life cycle, small size, and high transformation efficiency. This protocol outlines a replicable methodology for A. tumefaciens–mediated transformation of Micro-Tom from cotyledon explants, utilizing cost-effective plant growth regulators for shoot regeneration, high transformation rates, reduced regeneration time, and enhanced rooting conditions.

Optimized Secretome Sample Preparation From High Volume Cell Culture Media for LC–MS/MS Proteomic Analysis
适用于 LC–MS/MS 蛋白质组学分析的大体积细胞培养上清分泌组样品制备方法优化
The cellular secretome is a rich source of biomarkers and extracellular signaling molecules, but proteomic profiling remains challenging, especially when processing culture volumes greater than 5 mL. Low protein abundance, high serum contamination, and sample loss during preparation limit reproducibility and sensitivity in mass spectrometry–based workflows. Here, we present an optimized and scalable protocol that integrates (i) 50 kDa molecular weight cutoff ultrafiltration, (ii) spin column depletion of abundant serum proteins, and (iii) acetone/TCA precipitation for protein recovery. This workflow enables balanced recovery of both low- and high-molecular-weight proteins while reducing background from serum albumin, thereby improving sensitivity, reproducibility, and dynamic range for LC–MS/MS analysis. Validated in human mesenchymal stromal cell cultures, the protocol is broadly applicable across diverse cell types and experimental designs, making it well-suited for biomarker discovery and extracellular proteomics.
Vascularization of Human Pancreatic Islets With Adaptive Endothelial Cells for In Vitro Analysis and In Vivo Transplantation
利用适应性内皮细胞实现人胰岛体血管化,用于体外分析及体内移植
The pancreatic islet, the only type of tissue that secretes insulin in response to elevated blood glucose, plays a vital role in diabetes development and treatment. While various islet vascularization strategies have been developed, they have been hindered by major limitations such as relying on pre-patterning and the inability to span long distances. Furthermore, few strategies have demonstrated robust enough vascularization in vivo to support therapeutic subcutaneous islet transplantation. Using adaptive endothelial cells (ECs) reprogrammed by transient expression of the ETS Variant Transcription Factor 2 (ETV-2) gene, we have physiologically vascularized human islets within a generic microchamber and have achieved functional engraftment of human islets in the subcutaneous space of mice. Such adaptive ECs, which we term reprogrammed vascular ECs (R-VECs), have been proven to be a suitable tool for both in vitro disease modeling and in vivo transplantation of not only islets but also other organoids.
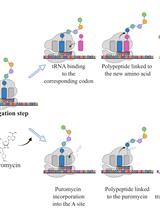
SUrface SEnsing of Translation (SUnSET), a Method Based on Western Blot Assessing Protein Synthesis Rates in vitro
翻译表面检测 (SUnSET),一种基于蛋白质印迹的体外蛋白质合成率评估方法
As the most energy- and metabolite-consuming process, protein synthesis is under the control of several intrinsic and extrinsic factors that determine its fine-tuning to the cellular microenvironment. Consequently, variations in protein synthesis rates occur under various physiological and pathological conditions, enabling an adaptive response by the ce•ll. For example, global protein synthesis increases upon mitogenic factors to support biomass generation and cell proliferation, while exposure to low concentrations of oxygen or nutrients require translational repression and reprogramming to avoid energy depletion and cell death. To assess fluctuations in protein synthesis rates, radioactive isotopes or radiolabeled amino acids are often used. Although highly sensitive, these techniques involve the use of potentially toxic radioactive compounds and require specific materials and processes for the use and disposal of these molecules. The development of alternative, non-radioactive methods that can be easily and safely implemented in laboratories has therefore been encouraged to avoid handling radioactivity. In this context, the SUrface SEnsing of Translation (SUnSET) method, based on the classical western blot technique, was developed by Schmidt et al. in 2009. The SUnSET is nowadays recognized as a simple alternative to radioactive methods assessing protein synthesis rates.Key features• As a structural analogue of aminoacyl-transfer RNA, puromycin incorporates into the elongating peptide chain.• Detection of puromycin-labeled peptides by western blotting reflects translation rates without the need for radioactive isotopes.• The protocol described here for in vitro applications is derived from the SUnSET method originally published by Schmidt et al. (2009).






