

Identifying and Characterizing Lipid-Binding Cavities in Lipid Transfer Proteins With CG-MD Simulations
基于粗粒化分子动力学模拟鉴定并表征脂质转运蛋白中的脂质结合腔
(*contributed equally to this work) 发布: 2025年12月20日第15卷第24期 DOI: 10.21769/BioProtoc.5558 浏览次数: 870
评审: Anonymous reviewer(s)
参见作者原研究论文
The authors used this protocol in:

Nov 2024
Advertisement










Abstract
Understanding how lipids interact with lipid transfer proteins (LTPs) is essential for uncovering their molecular mechanisms. Yet, many available LTP structures, particularly those thought to function as membrane bridges, lack detailed information on where their native lipid ligands are located. Computational strategies, such as docking or AI-methods, offer a valuable alternative to overcome this gap, but their effectiveness is often restricted by the inherent flexibility of lipid molecules and the lack of large training sets with structures of proteins bound to lipids. To tackle this issue, we introduce a reproducible computational pipeline that uses unbiased coarse-grained molecular dynamics (CG-MD) simulations on a free and open-source software (GROMACS) with the Martini 3 force-field. Starting from a configuration of a lipid in bulk solvent, we run CG-MD simulations and observe spontaneous binding of the lipid to the protein. We show that this protocol reliably identifies lipid-binding pockets in LTPs and, unlike docking methods, suggests potential entry routes for lipid molecules with no a priori knowledge other than the protein’s structure. We demonstrate the utility of this approach in investigating bridge LTPs whose internal lipid-binding positions remain unresolved. Altogether, our study provides a cost-effective, efficient, and accurate framework for mapping binding sites and entry pathways in diverse LTPs.
Key features
• Demonstrates the reliability of unbiased coarse-grain molecular dynamics (CG-MD) simulations with the Martini 3 force-field in identifying lipid-binding sites in lipid transfer proteins (LTPs).
• The protocol is straightforward to replicate, relying solely on freely available open-source software.
• Furthermore, it is computationally efficient, with most simulations completing within a few hours on a standalone GPU-accelerated workstation.
• As input, the user only needs to include the structure of the protein and select the lipid type to test.
Keywords: Coarse-grain MD simulations (粗粒化分子动力学模拟)Graphical overview
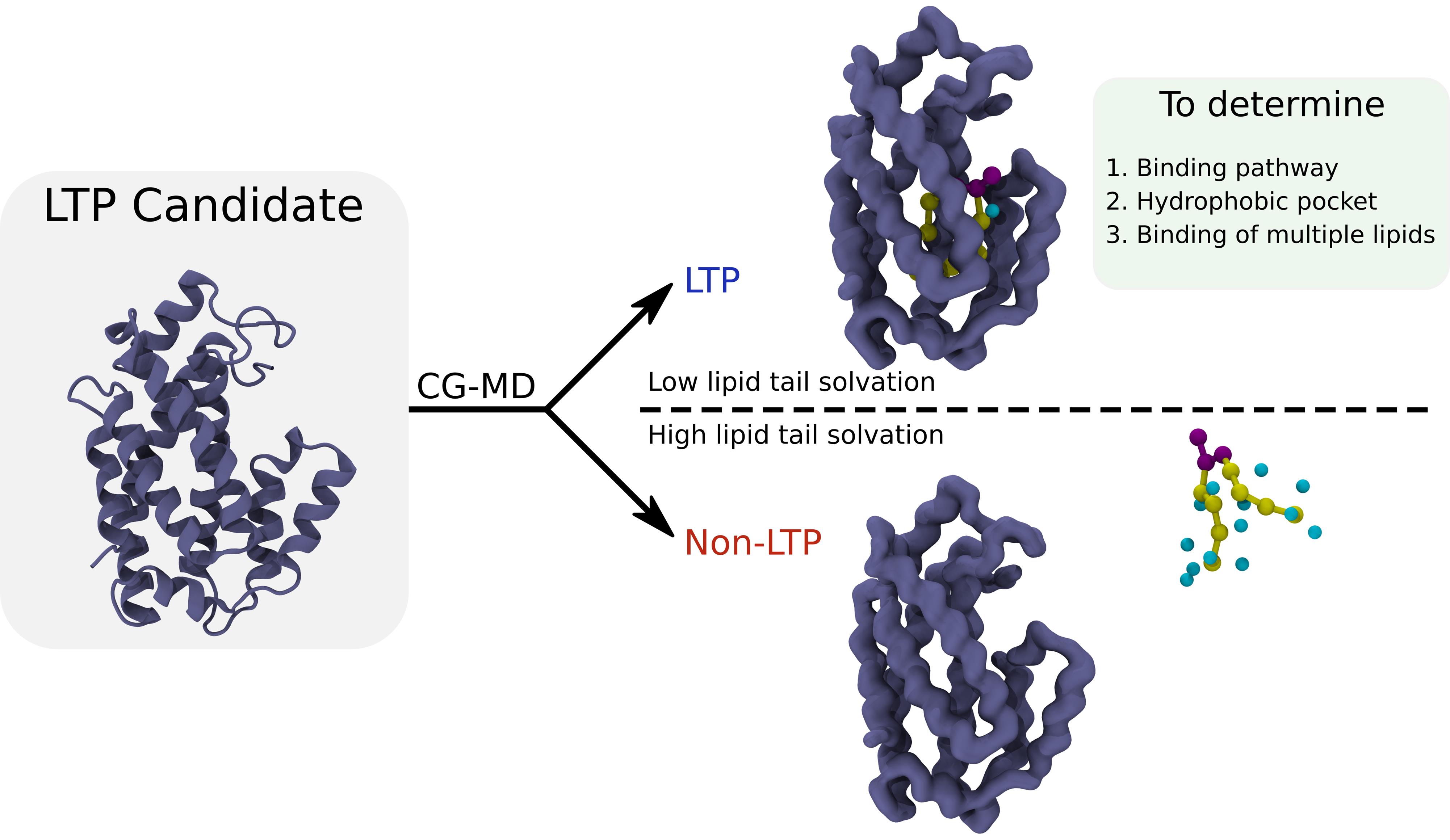
Coarse-grained molecular dynamics (CG-MD) simulations can identify the binding cavity of lipid transfer proteins. The protocol can also be used to characterize the lipid-binding cavity, as well as the binding pathway of the lipids. Further, it can be employed iteratively to study the binding of multiple lipids to the same cavity.
Background
Eukaryotic cells are compartmentalized into membrane-bound organelles, each defined by a unique lipid composition essential for its function. Preserving this lipid balance requires selective transport between organelles, mediated by both vesicular and non-vesicular mechanisms. A major class of mediators of non-vesicular lipid trafficking is lipid transfer proteins (LTPs), which encapsulate lipids in hydrophobic cavities and transfer them between membranes. In recent years, many new LTPs have been identified, highlighting the widespread role of this process in cellular physiology. Some LTPs are thought to operate at membrane contact sites, forming extended hydrophobic tunnels that physically bridge organelles and enable bulk lipid transfer.
Although significant progress has been made, the molecular mechanisms by which LTPs function remain only partially understood. Key challenges include determining how LTPs recognize donor and acceptor membranes with high specificity, and how they overcome energy barriers to extract and release lipids and achieve transport directionality. Structural biology has provided valuable insights, yet many high-resolution LTP structures lack bound lipids, likely reflecting their dynamic behavior within the binding cavity. For bridge-like LTPs (BLTPs) in particular, structural models often rely on AlphaFold predictions, leaving uncertainties about lipid positioning and transport pathways. These proteins are a recently discovered type of LTPs that are large enough to span the whole distance between organelle membranes at membrane contact sites (MCS), consisting of a long hydrophobic cavity that is able to accommodate tens of lipids simultaneously.
Computational approaches offer complementary tools to address these gaps. Traditional methods, such as molecular docking, are limited by the high flexibility of lipid substrates, while atomistic molecular dynamics (MD) simulations, although informative, are computationally demanding and not easily applied to large-scale studies. AI tools are also emerging as a popular tool for protein–ligand predictions, but they are untested in the case of lipids. Recent advances in coarse-grained molecular dynamics (CG-MD) simulations provide a promising alternative, allowing protein–ligand interactions to be captured with reduced computational cost [1]. In this sense, despite the lack of flexibility of the protein structure on CG-MD simulations, the hydrophobic character of the lipid-binding cavity allows reproducing the experimental binding of lipids with no false positives. More importantly, the dynamic view that MD simulations provide allows analyzing aspects of the lipid-binding process that cannot be inferred by docking or AI methods, such as the pathway for lipid entry.
In our recent publication [2], we adapted unbiased CG-MD simulations to investigate lipid–LTP interactions. By placing lipids in the solvent, we observed their spontaneous binding to LTPs, enabling the identification of binding pockets, entry pathways, and lipid density distributions within these proteins. Applying this protocol to bridge-like LTPs such as Vps13 and Atg2 revealed structural features of their lipid-binding activity that have remained unresolved experimentally. Our approach thus provides an efficient, accurate, and scalable framework to dissect the mechanisms of lipid transport and to guide the discovery of new LTP functions.
Equipment
The hardware and software requirements to run this protocol are a macOS or 64-bit Linux-based operating system, 8 GB of RAM, 40 GB of disk space, and a GPU compatible with GROMACS. We recommend using NVIDIA CUDA support whenever possible and building GROMACS in single precision mode. The commands used in this article were validated on Ubuntu 22.04 LTS.
We have tested the protocol in a workstation with a NVIDIA 4070 Ti GPU with CUDA version 12.2 and GROMACS 2023, testing an LTP of 70 kDa. Each replica of 1 μs takes 5 h to complete, with a performance of 4,800 ns/day. Therefore, the results from 5 replicas take around 1 day when run sequentially.
Software and datasets
1. Preparation of model: Martinize2 [3], 0.15.0, 2025, Apache 2.0, free
2. Preparation of model: DSSP[4], 3.0.0, 2018, boost, free
3. Simulations and Analysis: GROMACS [5], 2021.2, 2021, LGPL, free
4. Visualization and Analysis: VMD [6], 1.9.4a55, 2021, UIUC open source, free
All bash and TCL scripts for simulation and analysis are provided in the GitHub repository: https://github.com/danialv4/Unbiased_simulations_characterize_lipid_binding/tree/main/Workflow/common.
Martinize2 can be downloaded and installed from https://github.com/marrink-lab/vermouth-martinize.
GROMACS can be installed from https://ftp.gromacs.org/gromacs/gromacs-2025.3.tar.gz.
The instructions for all tools employed in this protocol are explained in the following section.
Procedure
文章信息
稿件历史记录
提交日期: Oct 3, 2025
接收日期: Nov 16, 2025
在线发布日期: Dec 9, 2025
出版日期: Dec 20, 2025
版权信息
© 2025 The Author(s); This is an open access article under the CC BY-NC license (https://creativecommons.org/licenses/by-nc/4.0/).
如何引用
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Álvarez, D., Vanni, S. and Srinivasan, S. (2025). Identifying and Characterizing Lipid-Binding Cavities in Lipid Transfer Proteins With CG-MD Simulations. Bio-protocol 15(24): e5558. DOI: 10.21769/BioProtoc.5558.
- Srinivasan, S., Álvarez, D., John Peter, A. T. and Vanni, S. (2024). Unbiased MD simulations identify lipid binding sites in lipid transfer proteins. J Cell Biol. 223(11): e202312055. https://doi.org/10.1083/jcb.202312055
分类
生物信息学与计算生物学
生物物理学 > 大分子模拟
您对这篇实验方法有问题吗?
在此处发布您的问题,我们将邀请本文作者来回答。同时,我们会将您的问题发布到Bio-protocol Exchange,以便寻求社区成员的帮助。