

Computational Workflow for Genome-Wide DNA Methylation Profiling and Differential Methylation Analysis
全基因组DNA甲基化分析与差异甲基化分析的计算流程


发布: 2025年11月05日第15卷第21期 DOI: 10.21769/BioProtoc.5506 浏览次数: 2335
评审: Migla MiskinyteAnonymous reviewer(s)
参见作者原研究论文
The authors used this protocol in:

Jan 2023
Advertisement








Abstract
DNA methylation is a crucial epigenetic modification that influences gene expression and plays a role in various biological processes. High-throughput sequencing techniques, such as bisulfite sequencing (BS-seq) and enzymatic methyl sequencing (EM-seq), enable genome-wide profiling of DNA methylation patterns with single-base resolution. In this protocol, we present a bioinformatics pipeline for analyzing genome-wide DNA methylation. We outline the step-by-step process of the essential analyses, including quality control using FASTQ for BS- and EM-seqs raw reads, read alignment with commonly used aligners such as Bowtie2 and BS-Seeker2, DNA methylation calling to generate CGmap files, identification of differentially methylated regions (DMRs) using tools including MethylC-analyzer and HOME, data visualization, and post-alignment analyses. Compared to existing workflows, this pipeline integrates multiple steps into a single protocol, lowering the technical barrier, improving reproducibility, and offering flexibility for both plant and animal methylome studies. To illustrate the application of BS-seq and EM-seq, we demonstrate a case study on analyzing a mutant in Arabidopsis thaliana with a mutation in the met1 gene, which encodes a DNA methyltransferase, and results in global CG hypomethylation and altered gene regulation. This example highlights the biological insights that can be gained through systematic methylome analysis. Our workflow is adaptable to any organism with a reference genome and provides a robust framework for uncovering methylation-associated regulatory mechanisms. All scripts and detailed instructions are provided in GitHub repository: https://github.com/PaoyangLab/Methylation_Analysis.
Key features
• Provides a comprehensive pipeline for genome-wide DNA methylation analysis.
• Step-by-step command line for DMR identification and post-analysis with visualization.
Keywords: Bisulfite sequencing (亚硫酸氢盐测序)Graphical overview
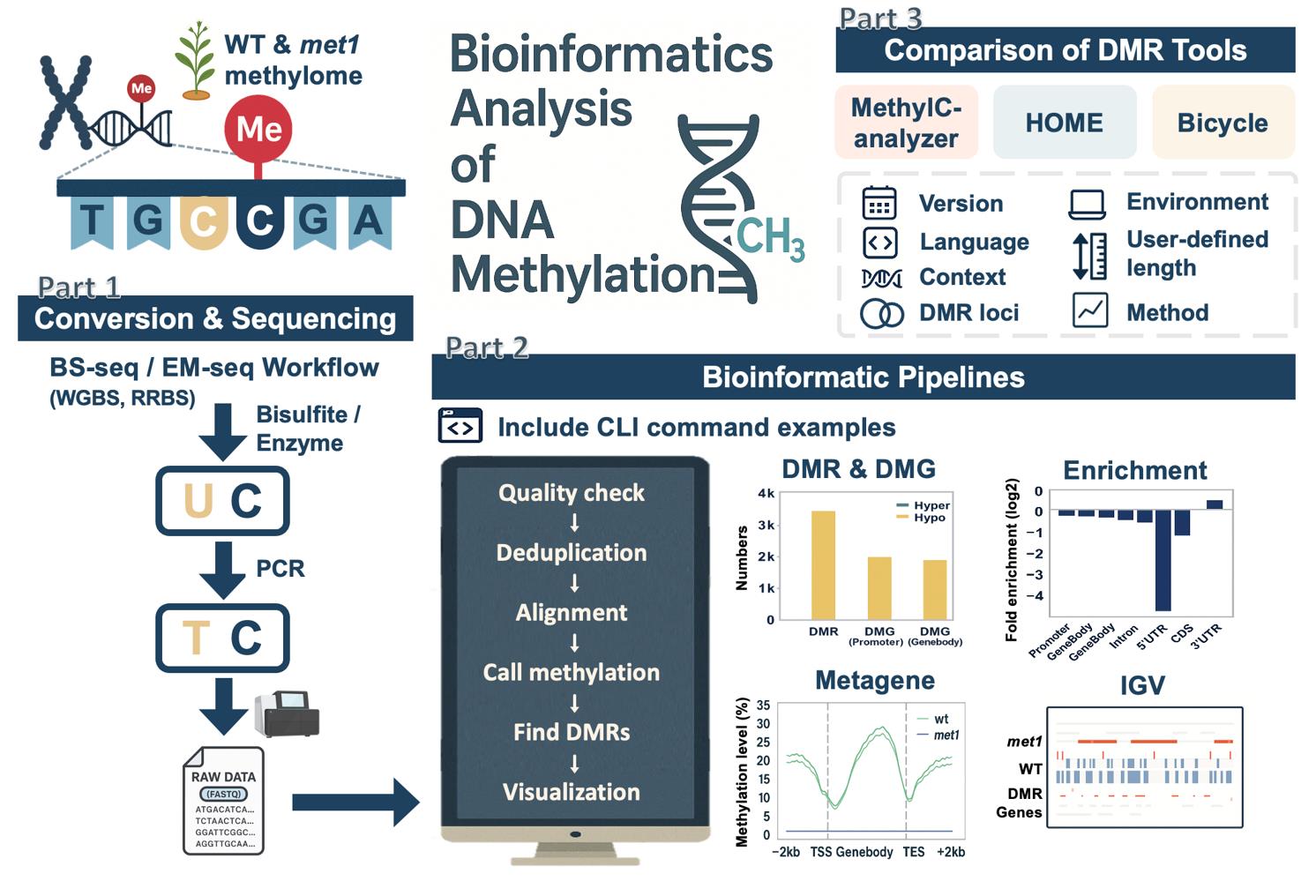
Overview of bioinformatics analysis of DNA methylation. An overview of this workflow, including the conversion and sequencing processes of bisulfite sequencing (BS-seq) and enzymatic methyl sequencing (EM-seq), the downstream bioinformatics workflow with example commands, and post-alignment analyses. It includes a comparison of three differentially methylated region (DMR) identification tools, highlighting differences in methodology, feature support, and DMR outputs.
Background
Epigenetics refers to alterations in gene expression that do not involve any change in the underlying DNA sequences. Such modifications can be inherited and are often reversible [1]. Among all epigenetic factors, DNA methylation is the most studied epigenetic regulator; it refers to the mechanism by which a methyl group is transferred to the C5 position of cytosine to form 5-methylcytosine (5mC) via DNA methyltransferases (DNMTs). DNA methylation occurs in the contexts of symmetric CG and CHG as well as asymmetric CHH sites, where H represents A, C, or T. In symmetric contexts (CG and CHG), methylation can be maintained across DNA replication because the complementary strand provides a template for restoring the methylation pattern. In contrast, methylation at asymmetric CHH sites lacks such symmetry and therefore requires continuous de novo establishment by specific DNA methyltransferases, making it more dynamic and often associated with transposon silencing.
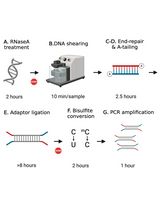
DNA methylation can silence genes or transposable elements by changing the chromatin structure or interfering with transcription factor binding [2]. Due to the importance of DNA methylation in biological processes, experimental approaches have been developed to profile genome-wide DNA methylation. Genome-wide DNA methylation profiling is commonly performed using next-generation sequencing (NGS) methods such as reduced-representation bisulfite sequencing (RRBS) [3], whole-genome bisulfite sequencing (WGBS) [4], and enzymatic methyl sequencing (EM-seq) [5]. These NGS-based approaches can determine the methylation status of DNA sequences at single-base resolution and measure DNA methylation levels digitally. In bisulfite sequencing (BS) methods such as RRBS, BS-seq, or WGBS, the crucial step is sodium bisulfite treatment, which converts unmethylated cytosine to uracil (and then to thymine during PCR), while 5mC remains unchanged (Figure 1A). Such treatment can result in approximately 84%–96% DNA degradation, causing a significant loss of DNA material and induction of sequence bias, therefore affecting the accuracy of the analyses [6]. To improve from the bisulfite treatment in BS-seq, EM-seq is performed to reduce DNA damage and produce higher-quality libraries for detecting 5mC, and it can generate comparable results using as little as 0.5 ng of input DNA compared with the 200 ng typically required for BS-seq, representing approximately a 400-fold reduction in input material. It uses two sets of enzymatic reactions, methylcytosine dioxygenase 2 (TET2) and T4-phage beta-glucosyltransferase (T4-BGT), to convert 5mC and 5hmC into products that cannot be deaminated by apolipoprotein B mRNA editing enzyme catalytic subunit 3A (APOBEC3A). Then, APOBEC3A deaminates unmodified C to generate U, which is eventually converted into T during PCR (Figure 1A) before the final library is sequenced. Compared to BS-seq, EM-seq offers a higher yield and better genome coverage with fewer PCR cycles required [7]. Unlike bisulfite libraries, EM-seq libraries do not exhibit biased AT-rich, GC-poor sequence representation due to the absence of bisulfite treatment–induced DNA damage [5]. Moreover, low-input EM-seq libraries provide similar results to high-input libraries; for instance, a 0.5 ng input of EM-seq covers more CpGs (regions of DNA where a cytosine is followed by a guanine) than the 200 ng input used in BS-seq, highlighting the higher sensitivity of EM-seq [5].
Profiling genome-wide DNA methylation can be computationally intensive [8,9]. The general workflow for such bioinformatics analysis usually includes assessment of read quality, removal of duplicated reads, alignment of reads, quantification of DNA methylation levels, identification of differentially methylated regions (DMRs), visualization of the methylome, and other post-alignment analyses (Figure 1B). Each step involves handling large sequencing datasets, often tens to hundreds of gigabytes per sample, which requires substantial computational resources. Alignment of bisulfite-treated reads is particularly demanding due to reduced sequence complexity, and parallel computing is often necessary to achieve reasonable runtimes. Memory usage ranges from 8 to 16 GB for small genomes such as Arabidopsis thaliana, but analyses of mammalian WGBS typically require more. Disk space is also a major consideration, with at least 2 TB of available storage being advisable for medium-scale projects.
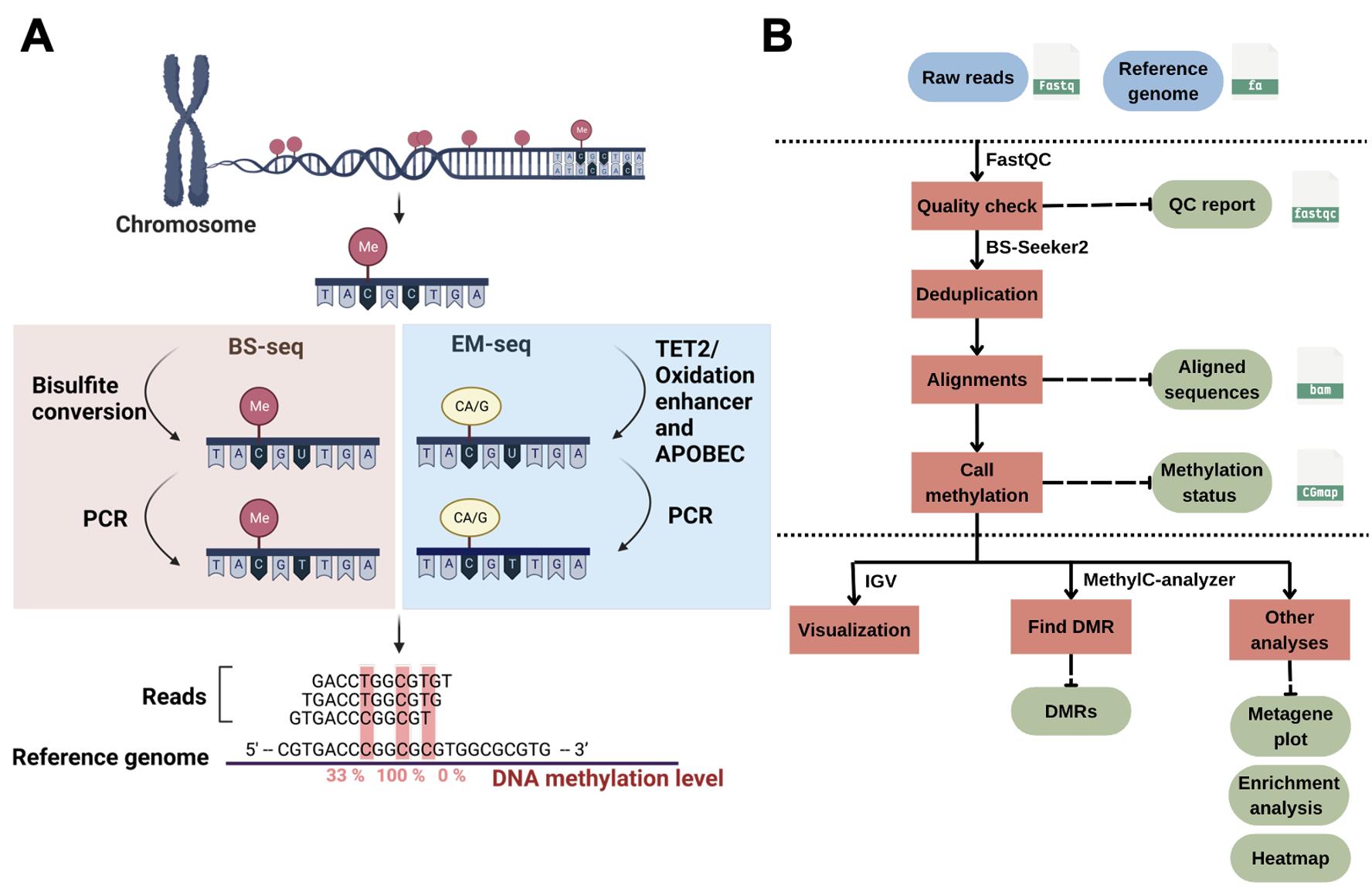
Figure 1. DNA methylation and bioinformatics pipelines overview. (A) Library construction of the enzymatic methyl sequencing (EM-seq) and bisulfite sequencing (BS-seq). Created using BioRender (http://biorender.com/). (B) Workflow of bioinformatics pipelines for DNA methylation analysis. The blue color represents the input data, while the green color is the output. Red boxes are the steps for analysis, and the suggested tools are listed above the box.
Read alignment
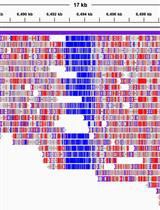
Aligning reads to the reference genome is a critical first step in identifying methylated DNA sites from DNA methylation sequencing data. Commonly used bisulfite-read aligners can carry it out with two types of algorithms: wild-card aligners [10] and three-letter aligners [11]. Wild-card aligners, such as BSMAP [12], replace Cs in the reference genome with the wild-card letter Y, which can match both Cs and Ts in the bisulfite-converted reads. This method offers higher genomic coverage, but can also introduce a bias toward higher methylation levels since it reduces the ability to distinguish between truly methylated cytosines (retained as C) and unmethylated cytosines that have been converted to T. As a result, reads containing Ts may be misaligned to C positions in the reference, leading to an overestimation of methylated sites [11]. On the other hand, three-letter aligners, such as Bismark [13], BS-Seeker2 [14], and BS Seeker3 [15], have higher mapping accuracy but slightly lower coverage compared to the wild-card aligners [11], as they convert all Cs into Ts in the reads for both strands of the sequence, resulting in lower mapping complexity. In general, Bismark is more accurate than BSMAP but its mapping rate and accuracy may decrease with high read error rates in longer reads [16]. BS-Seeker2 is more capable of mapping reads from problematic libraries [14], and is only minimally impacted by read error rates [16]. Overall, among these tools, BSMAP offers the fastest alignment speed and minimal memory usage, while BS-Seeker2 provides the highest mapping accuracy [17]. Also, BS-Seeker3 [15] is an updated version of BS-Seeker2 that enhances alignment accuracy and mappability, while reducing computational time. Common issues during read alignment, like low conversion rate (<98%), may affect accurate cytosine calling and lead to underestimated conversion efficiency. Removing low-quality reads using quality control tools before proceeding can avoid such potential problems. Aligners output the alignments as BAM or SAM files [18] and methylation calling results of each C base as CGmap files [19]; each row contains CpG site information, including chromosome, strand, genomic position, sequence context (CG, CHG, or CHH), dinucleotide context, methylation level, and read counts (Figure 2).
Cytosine methylation–level information from CGmap files can be utilized for identifying DMRs. These refer to genomic regions with significantly different levels of DNA methylation between two groups of methylomes—complete sets of DNA methylation patterns across the genome of an organism, cell type, or condition (e.g., experimental and control). The locations of DMRs may be further linked to specific, biologically meaningful features, such as promoters, genes, CpG islands, or other user-defined regions [18,19]. In general, CG sites within promoters often show consistent hypomethylation across biological replicates, correlating with transcriptional activity, whereas intergenic CHH sites are more variable and context-dependent. Additionally, individual variation and potential bias introduced by bisulfite sequencing may contribute to subtle differences between biological replicates, which therefore share similar but not identical methylomes (methylation profiles).
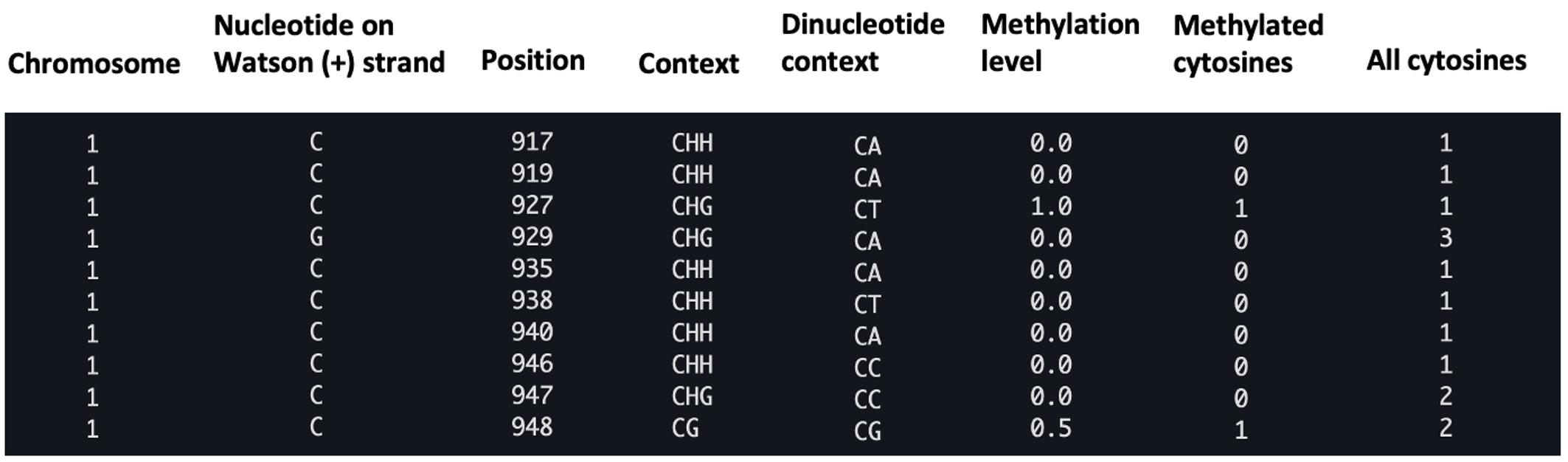
Figure 2. Example of a CGmap file generated with the BS-seeker2 call methylation script. The figure displays a screenshot of the ten rows from the “wt_r1.CGmap.gz” file.
Differential methylation region (DMR) identification
Several tools have been developed for DMR detection. Here, we highlight three popular ones, HOME [20], MethylC-analyzer [21], and Bicycle [22] (Table 1), each based on different approaches. Machine learning methods make few distributional assumptions and can capture nonlinear patterns. By implementing the machine learning algorithm, HOME utilizes a trained support vector machine (SVM) model to score each cytosine by specific features computed by weighted logistic regression using methylation level differences and p-values between two groups. The tool groups cytosines into DMRs based on scores and distances to their neighboring cytosines [20]. The prebuilt SVM model in HOME has primarily been designed for analyzing mammalian (mainly human) DNA methylation data; therefore, it incorporates assumptions that may not account for the unique genetic regulation in nonmammalian species [23]. DMRs found by HOME are predicted by a precise delineation of the boundaries, and the lengths of the DMRs can vary widely.
Statistical DMR identification tools, such as the MethylC-analyzer, identify DMRs by comparing the average methylation levels (Δ methylation) of the genomic regions between the two groups. A larger difference (e.g., ≥10%) suggests a more substantial shift in epigenetic state. It also offers users a choice between three statistical methods, the Student’s t-test, the Kolmogorov–Smirnov test, and the Mann–Whitney U test, for detecting DMRs with significant differences [21] that reflect that the variation is consistent across replicates, instead of occurring by chance. These statistical methods apply classical hypothesis tests to predefined regions or windows, offering simplicity and speed but inheriting test assumptions (e.g., approximate normality, independent observations) [24] that may be affected when sample sizes are small [25]. Additionally, although users can customize the length of DMRs in MethylC-analyzer, fixed-length regions may still miss irregular DMR boundaries.
As a model-based DMR-finding tool, Bicycle compares methylation levels of user-defined regions between two groups and identifies DMRs using the likelihood ratio test based on beta-binomial models with considerations for sensitivity and specificity [22]. Using beta-binomial models has been claimed to decrease the false-positive rate in DMR identification. In brief, tool selection can be based on data type and analysis requirements, as different tools employ different approaches to define DMRs with diverse lengths and characteristics (see Table 1 for an overview of these tools).
Table 1. Comparison of three DMR tools
| Feature/tool | HOME | MethylC-analyzer | Bicycle |
|---|---|---|---|
| Version | 1.0.0 | - | 1.8.2 |
| Language | Python, R | Python, R | java |
| Environment | CLI/ | CLI/Docker | CLI |
| Available context | CG, CHG, CHH | CG, CHG, CHH | CG, CHG, CHH |
| Testing method | Weighted logistic regression, support vector machine | Student’s t-test, Kolmogorov–Smirnov test, Mann–Whitney U test | Likelihood ratio of beta-binomial models |
| User-defined DMR length | not available | available | available |
CLI: command line interface, GUI: graphical user interface
Data visualization
After the reads are aligned, methylome data can be visualized by Integrative Genomic Viewer (IGV) [26] or the UCSC Genome Browser [27]. Users can customize the tracks on both the IGV and UCSC Genome Browser for a better understanding of the global DNA methylation pattern and compare it with other genome features ranging from single-nucleotide to megabase scales. IGV is a user-friendly desktop application that allows users to visualize methylation sites on the genome easily by importing files such as wiggle (WIG) files [27], a text-based format file that stores quantitative genomic data, such as methylation levels or read coverage, across genomic coordinates for efficient visualization. With IGV, we can directly view the methylation levels of identified DMRs and explore the adjacent genomic region that may be the potential regulatory targets of identified DMRs.
Post-alignment analyses
Post-alignment analyses aim to associate genomic regions with identified DMRs and explore the roles of these DMRs in genomic regulatory mechanisms, where various toolkits can be applied to such analyses. The R package methylKit [28] can identify DMR proportions in various genetic elements, such as promoters, exons, or enhancers. MethGO [29] provides several modules for analyzing the correlation between methylation level and genomic features, including transcription factor-binding sites (TFBSs). MethylC-analyzer [21] provides an easy-to-use pipeline following the DMR identification step and includes several common analyses, such as enrichment analysis and metagene analysis. Enrichment analysis can assess the preferential localization of DMRs within genomic features across the genome, and metagene analysis is able to show the distribution of methylation levels along the gene body and adjacent regions. MeH is another useful tool that estimates methylation heterogeneity within a population of cells and allows investigation of intra-sample methylation dynamics at specific genomic loci [30].
Software and datasets
All scripts, examples, and software information (Table 2) are provided in the README at our GitHub repository: https://github.com/PaoyangLab/Methylation_Analysis.
Table 2. Required software tools, datasets, and resources used in this pipeline.
| Software/dataset/resource | Version | Date | Access | Dependencies |
|---|---|---|---|---|
| SRA Toolkit [31] | v3.0.5 | May 9, 2023 | Open access | |
| FastQC [32] | v0.12.0 | March 1, 2023 | Open access | |
| TrimGalore [33] | v0.6.10 | Feb 2, 2023 | Open access | |
| Bowtie2 [34] | v2.26 | August 24, 2015 | Open access | C/C++ libraries |
| BS-Seeker2 [14] | v2.1.8 | October 31, 2018 | Open access | Python ≥ 2.7 |
| HOME v1.0.0 [20] | v1.0.0 | February 4, 2019 | Open access | |
| MethylC-analyzer [21] | - | January 6, 2023 | Open access | Docker ≥ v20.10 |
| Bicycle [22] | v1.8.2 | April 25, 2020 | Open access | |
| IGV Desktop [26] | v2.16.0 | April 19, 2023 | Open access | |
| GSE122394 [35] | - | November 20, 2019 | Open access |
Genome-wide DNA methylation dataset
To demonstrate the methylation analysis pipeline, we downloaded and processed Arabidopsis thaliana (GSE122394) BS-seq datasets [35], including wild-type (wt) strains as controls and met1 mutant strains. The met1 mutants lack functional DNA methyltransferase 1 (MET1), which is primarily responsible for maintaining CG methylation [36]. Each group contained three biological replicates.
Data for project GSE122394 are available on Gene Expression Omnibus (GEO) and can be accessed using the provided accession codes. The raw reads for each sample are stored in the Sequence Read Archive (SRA) listed in the GEO. To obtain the data, you can use SRAToolkit [33] to download the file using prefetch and then convert it into the FASTQ format (.fastq) for analysis by fast-dump. A FASTQ file is a text-based format that stores raw sequencing reads along with their corresponding base quality scores from high-throughput sequencing experiments.
Note: fasterq-dump is faster by using multiple threads but requires substantial temporary disk space (approximately 3–4 times the size of the SRA file).
prefetch SRR8180314 ## download SRA datafast-dump SRR8180314 ## transfer into fastq filemv SRR8180314.fastq wt_r1.fastq ## rename the file # Convert SRA file into FASTQ using fasterq-dump (multi-threaded, more efficient)# fasterq-dump SRR8180314 --split-files -e 8 -t ./tmpHardware requirements
We recommend a workstation with 8–16 CPU cores, 32–64 GB RAM, and at least 1.5 TB of storage. Raw FASTQ files of Arabidopsis typically require 80–120 GB per sample, with BAM and CGmap outputs adding ~40–70 GB each. A dataset of six samples usually needs ~1 TB in total, but allocating ≥2 TB (preferably SSD) ensures smooth analysis.
Procedure
文章信息
稿件历史记录
提交日期: Jul 24, 2025
接收日期: Oct 10, 2025
在线发布日期: Oct 22, 2025
出版日期: Nov 5, 2025
版权信息
© 2025 The Author(s); This is an open access article under the CC BY license (https://creativecommons.org/licenses/by/4.0/).
如何引用
Lin, P. Y., Lin, G. J., Chen, K. L., Huang, S. C. and Chen, P. Y. (2025). Computational Workflow for Genome-Wide DNA Methylation Profiling and Differential Methylation Analysis. Bio-protocol 15(21): e5506. DOI: 10.21769/BioProtoc.5506.
分类
生物信息学与计算生物学
系统生物学 > 表观基因组学 > DNA 甲基化
系统生物学 > 基因组学
您对这篇实验方法有问题吗?
在此处发布您的问题,我们将邀请本文作者来回答。同时,我们会将您的问题发布到Bio-protocol Exchange,以便寻求社区成员的帮助。