

Examining the Roles of m6A Sites in mRNA Using the Luciferase Gene Fused With Mutated RRACH Motifs
利用融合突变RRACH基序的荧光素酶基因研究mRNA中m6A位点的功能
发布: 2025年11月05日第15卷第21期 DOI: 10.21769/BioProtoc.5492 浏览次数: 2282
评审: Sébastien GillotinAnonymous reviewer(s)
参见作者原研究论文
The authors used this protocol in:

Apr 2025
Advertisement








Abstract
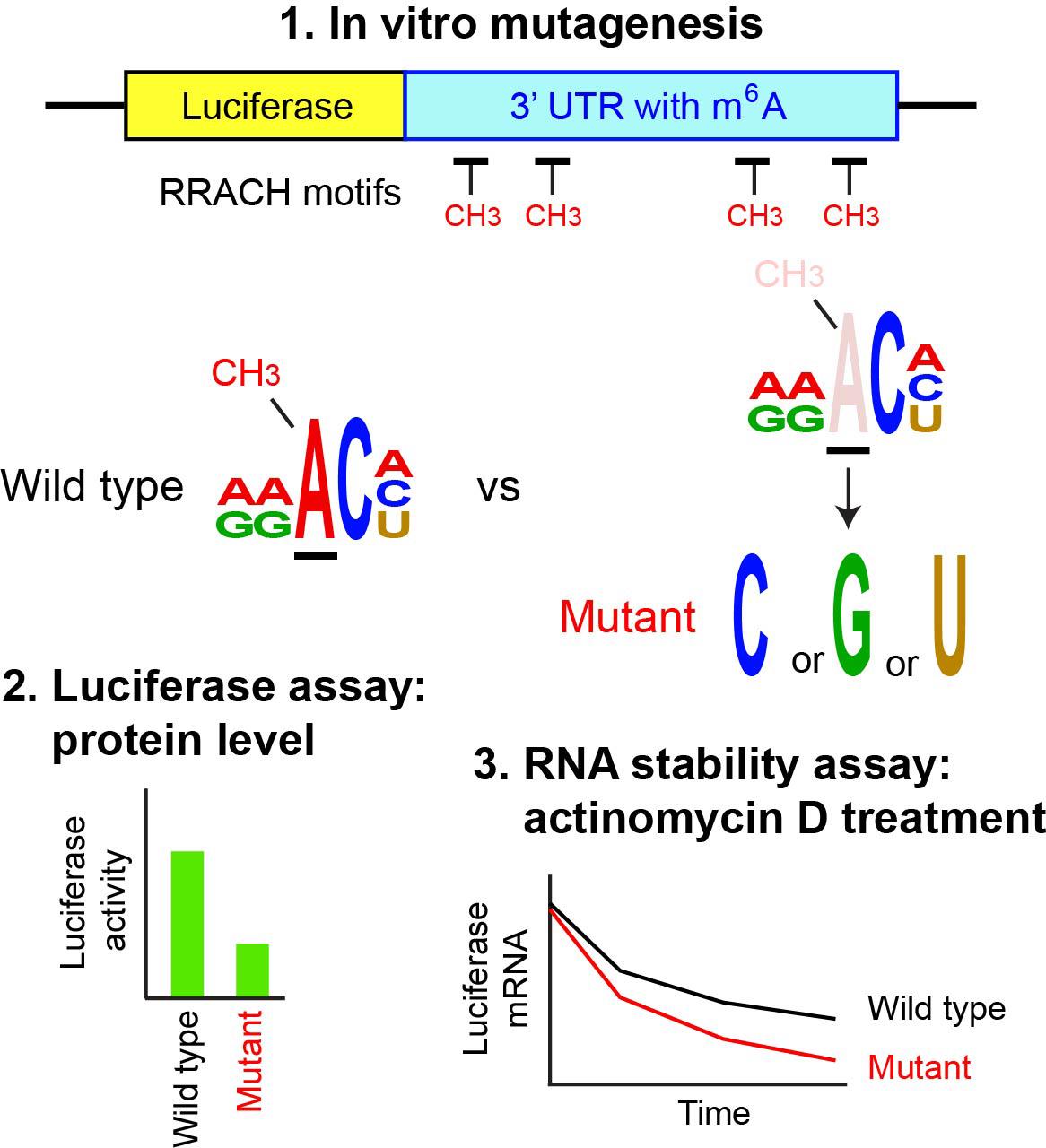
N6-methyladenosine (m6A) is the most abundant internal modification in mRNA and is regulated primarily by the balance between the METTL3 methylase complex and two demethylases, FTO (fat mass and obesity-associated protein) and ALKBH5 (α-ketoglutarate-dependent dioxygenase alkB homolog). Reflecting this prevalence, m6A participates in virtually every step of RNA metabolism, influencing a wide range of physiological and pathological processes. The first step in studying m6A is genome-wide mapping, typically performed by m6A-seq, which sequences RNA fragments immunoprecipitated with an m6A-specific antibody. This is followed by identification of RRACH motifs (R = A or G; H = A, C, or U) within these sequences, with m6A being located at the third nucleotide. The second step involves mutating the putative m6A sites to establish a causal link between the modification and downstream biological effects. Since the mapping step has been covered in several detailed protocols, this article focuses on the second step—mutagenesis of RRACH motifs and subsequent functional analysis of the mutations by ectopic expression. The 3′ untranslated region (UTR) of the mouse Runx2 gene is used as an example. The mutant and wild-type sequences are inserted into a luciferase reporter vector and transfected into 293FT cells to evaluate how loss of m6A affects luciferase protein levels. The same reporter plasmids are also used in an RNA stability assay with a transcription inhibitor. Although site-specific demethylation of endogenous mRNA would be preferable, it remains technically challenging despite many attempts. Thus, ectopic expression of the mutated target gene remains a widely used and practical alternative.
Key features
• This protocol assumes that the m6A-seq data and a list of putative target genes modified by m6A are already available.
• Highly specialized techniques or equipment are not required.
• The luciferase-based assessment in this protocol is widely used in the m6A field as a standard approach.
Keywords: Luciferase reporter (荧光素酶报告基因)Graphical overview
Background
m6A is the most abundant internal modification in mRNA and long non-coding RNA, among more than 150 known types of post-transcriptional RNA modifications [1–3]. m6A was first reported in 1974 [4,5], but the field remained relatively inactive until two key discoveries. First, in 2011, FTO (fat mass and obesity-associated protein) was identified as the first m6A demethylase, demonstrating the reversibility of this modification [6]. Interestingly, the m6A methylase METTL3 had been identified earlier, in 1994 [7]. Second, in 2012, two groups independently reported genome-wide mapping approaches, MeRIP-seq and m6A-seq, using an m6A-specific antibody in combination with next-generation sequencing [8,9]. Identification of additional relevant proteins soon established the principles governing m6A regulation and function. The core m6A methylase complex contains at least seven subunits, including METTL3 (catalytic subunit) and METTL14 (structural support essential for the METTL3 activity). m6A is typically found at the consensus RRACH sequence, often near stop codons and within 3′ UTR of mRNAs. The m6A level is dynamically regulated by the METTL3 complex (“writer”) and two demethylases (“erasers”), FTO and ALKBH5, during development and in response to environmental changes. m6A is recognized by “reader” proteins, such as the YTH domain family (YTHDC1–2, YTHDF1–3) and insulin-like growth factor 2-binding proteins (IGF2BPs), which influence RNA stability, splicing, nuclear transport, localization, translation, and RNA–protein interactions. Reflecting this versatility, m6A participates in diverse cellular processes, developmental events, and disease pathogenesis.
The following workflow represents a standard approach for studying the role of m6A in a specific mRNA within a given biological context. First, Mettl3 or Mettl14 is depleted by knockout (KO) or knockdown (KD) in cells or animals to remove m6A genome-wide, and the resulting phenotypes are described. Second, the genomic distribution of m6A is mapped with m6A-seq, which includes RNA-seq as the input. Third, mRNAs dysregulated at both the RNA and m6A levels following KO or KD are identified. Fourth, putative causal genes are determined by combining this list with literature evidence. The final and most critical step is proving a causal link between loss of m6A and the observed phenotype; however, this has been challenging. Since KO and KD effects may reflect the combined disruption of many genes, identifying a small subset sufficient to explain the phenotype requires site-specific demethylation of m6A. This, in turn, demands prior mapping at the single-nucleotide resolution, which m6A-seq cannot provide due to its use of RNA fragments up to ~200 bases. Although single-nucleotide mapping and site-specific demethylation techniques have been reported, the field is still developing (see references in [10]). A more widely used alternative, described here, is to transfect a luciferase reporter plasmid containing the putative m6A target sequence, with and without RRACH motif mutations, and measure luciferase activity as a surrogate for protein levels. The same reporter can also be used to examine how m6A affects RNA stability. While this ectopic expression approach is less direct than endogenous site-specific demethylation, it remains a practical and validated method, as demonstrated by several published examples listed at the end of this article. This protocol focuses on the final step, starting from the mutations of the adenine in the third nucleotide of each RRACH motif, assuming that m6A mapping data are already available. The RRACH motifs in the Runx2 gene are used as examples. The mRNA and protein of Runx2 are downregulated in the mouse limb bud cells of Mettl14 conditional KO (cKO) mice at embryonic day 12.5, accompanied by reduced m6A in the 3′ UTR [11]. For details on m6A-seq, readers are referred to comprehensive published protocols [12–14].
Materials and reagents
Biological materials
1. 293FT cells (Thermo Fisher Scientific, catalog number: R70007)
2. 293T/17 cells (ATCC, catalog number: CRL-11268)
Reagents
1. pGL3 control vector (Promega, catalog number: E1741, discontinued) or pGL4.13 vector (Promega, catalog number: E6881, replacement)
2. Renilla luciferase vector (pcDNA3.1dsRLuc) (Addgene, catalog number: 68053)
3. Gibson Assembly Master Mix (New England Biolabs, catalog number: E2611L)
4. Dulbecco’s modified Eagle medium (DMEM) (Corning, catalog number: 10-013-CM)
5. Opti-MEM (Thermo Fisher Scientific, catalog number: 31985070)
6. Fetal bovine serum (FBS) (Hyclone, catalog number: SH30071.03)
7. MEM non-essential amino acid, 10 mM (100×) (Thermo Fisher Scientific, catalog number: 11140050)
8. Geneticin (Thermo Fisher Scientific, catalog number: 10131035)
9. Lipofectamine 2000 transfection reagent (Thermo Fisher Scientific, catalog number: 11668027)
10. Actinomycin D (Sigma-Aldrich, catalog number: A1410)
11. Dimethyl sulfoxide (Thermo Fisher Scientific, catalog number: J66650.AK)
12. F-Luc forward primer, 5′-CGGAAAGACGATGACGGAAA-3′ (Thermo Fisher Scientific)
13. F-Luc reverse primer, 5′-CGGTACTTCGTCCACAAACA-3′ (Thermo Fisher Scientific)
14. Mouse 18S rRNA forward primer, 5′-AGTCCCTGCCCTTTGTACACA-3′ (Thermo Fisher Scientific)
15. Mouse 18S rRNA reverse primer, 5′-CGATCCGAGGGCCTCACTA-3′ (Thermo Fisher Scientific)
16. Dual-Luciferase Reporter Assay System (Promega, catalog number: E1910)
17. Click-iT RNA AlexaFluor 594 Imaging kit (Thermo Fisher Scientific, catalog number: C10330)
Solutions
1. Culture medium for 293FT cells (see Recipes)
Recipes
1. Culture medium for 293FT cells
| Component | Volume | Final concentration |
|---|---|---|
| DMEM | 440 mL | 88% |
| FBS | 50 mL | 10% |
| 10 mM (100×) MEM non-essential amino acid | 5 mL | 1× |
| 50 mg/mL geneticin | 5 mL | 500 μg/mL |
| Total | 500 mL | n/a |
Laboratory supplies
1. 24-well plates (Thermo Fischer Scientific, catalog number: 142485)
2. 96-well plates (Thermo Fischer Scientific, catalog number: 161093)
Equipment
1. Spectrophotometer (DeNovix, model: DS-11 FX+)
2. Luminometer (Beckman Coulter, model: LD 400)
Software and datasets
1. Integrative Genomics Viewer (IGV) (https://igv.org/)
2. Mouse genome assembly GRCm38/mm10 (https://www.ncbi.nlm.nih.gov/datasets/genome/GCF_000001635.20/)
3. NEBuilder Assembly Tool, version 2.10.8 (https://nebuilder.neb.com/#!/)
4. GraphPad Prism 10.1.0 (GraphPad)
Procedure
文章信息
稿件历史记录
提交日期: Aug 14, 2025
接收日期: Sep 26, 2025
在线发布日期: Oct 14, 2025
出版日期: Nov 5, 2025
版权信息
© 2025 The Author(s); This is an open access article under the CC BY-NC license (https://creativecommons.org/licenses/by-nc/4.0/).
如何引用
Katoku-Kikyo, N. and Kikyo, N. (2025). Examining the Roles of m6A Sites in mRNA Using the Luciferase Gene Fused With Mutated RRACH Motifs. Bio-protocol 15(21): e5492. DOI: 10.21769/BioProtoc.5492.
分类
分子生物学 > RNA > RNA 修饰
发育生物学 > 形态建成 > 器官形成
您对这篇实验方法有问题吗?
在此处发布您的问题,我们将邀请本文作者来回答。同时,我们会将您的问题发布到Bio-protocol Exchange,以便寻求社区成员的帮助。