

RACE-Nano-Seq: Profiling Transcriptome Diversity of a Genomic Locus
RACE-Nano-Seq:解析基因组特定位点的转录组多样性
发布: 2025年07月05日第15卷第13期 DOI: 10.21769/BioProtoc.5374 浏览次数: 2810
评审: Anonymous reviewer(s)
参见作者原研究论文
The authors used this protocol in:

Nov 2024
Advertisement









Abstract
The complexity of the human transcriptome poses significant challenges for complete annotation. Traditional RNA-seq, often limited by sensitivity and short read lengths, is frequently inadequate for identifying low-abundant transcripts and resolving complex populations of transcript isoforms. Direct long-read sequencing, while offering full-length information, suffers from throughput limitations, hindering the capture of low-abundance transcripts. To address these challenges, we introduce a targeted RNA enrichment strategy, rapid amplification of cDNA ends coupled with Nanopore sequencing (RACE-Nano-Seq). This method unravels the deep complexity of transcripts containing anchor sequences—specific regions of interest that might be exons of annotated genes, in silico predicted exons, or other sequences. RACE-Nano-Seq is based on inverse PCR with primers targeting these anchor regions to enrich the corresponding transcripts in both 5' and 3' directions. This method can be scaled for high-throughput transcriptome profiling by using multiplexing strategies. Through targeted RNA enrichment and full-length sequencing, RACE-Nano-Seq enables accurate and comprehensive profiling of low-abundance transcripts, often revealing complex transcript profiles at the targeted loci, both annotated and unannotated.
Key features
• This protocol is highly sensitive and can detect low-abundance transcripts.
• This protocol can be performed in a typical molecular biology laboratory.
• This protocol allows RACE reactions with single or multiple primers, supporting various research scales.
• This protocol enables characterization of complex genomic loci and discovery of novel transcripts, exons, and alternative splicing events.
Graphical overview
Background
The well-documented transcriptome complexity can be viewed as a combination of several hallmark features, including the multitude of alternative splicing events [1,2], multiple alternative transcription start and termination sites (TSSs and TTSs) [3,4], overlapping or chimeric transcripts [5–9], and pervasive transcription [10,11]. Collectively, the molecular processes that give rise to these features orchestrate the sophisticated landscape of mammalian gene expression.
Comprehensive characterization of complex transcriptome landscapes presents several challenges, particularly in detecting low-abundance transcripts, accurately defining transcript structures, and efficiently obtaining full-length sequences. While conventional transcriptome studies effectively identify and annotate highly expressed, ubiquitously present transcripts, they frequently miss elusive transcripts exhibiting tightly regulated expression patterns, produced only in specific tissues, cell types, or under specific biological conditions [12–16]. The restricted expression patterns of these transcripts result in extremely low abundance that often falls below the sensitivity limits of standard RNA-seq assays, hindering their effective detection.
Given these limitations, targeted RNA enrichment approaches have become essential for accessing low-abundance transcripts, especially in bulk samples. Currently, two primary targeted RNA enrichment strategies are employed: CaptureSeq and rapid amplification of cDNA ends (RACE). CaptureSeq, a hybridization-based method, utilizes specifically designed DNA oligonucleotide probes to capture transcripts of interest, thereby increasing sequencing coverage for target regions [7]. This method enhances the sensitivity and accuracy of low-abundance transcript detection and has been applied in the discovery of novel genes and transcripts [7,17]. Moreover, combining CaptureSeq with long-read sequencing platforms like Nanopore [18] or PacBio [19] enables high-precision sequencing without the need for transcript assembly. However, its implementation is costly and highly dependent on probe design accuracy.
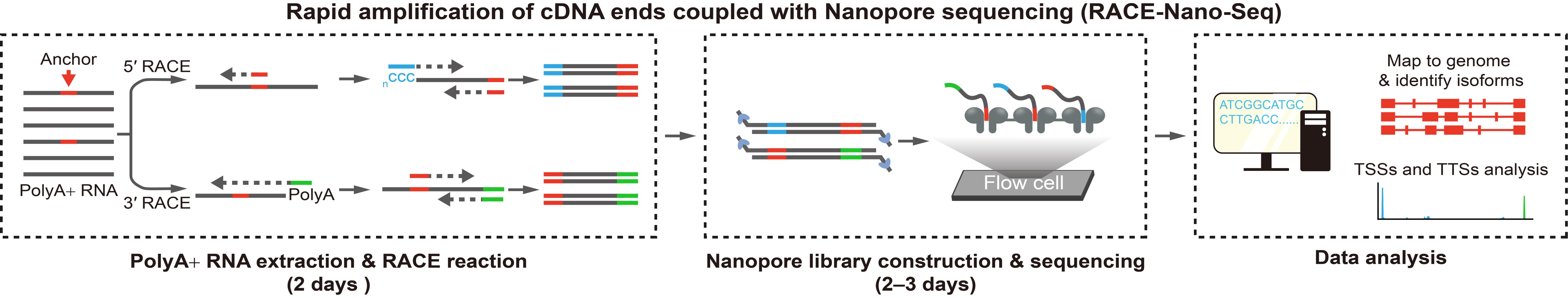
RACE, an inverse PCR-based technique, amplifies the 5' and 3' terminal sequences of RNA molecules by targeting specific anchor sequences [20,21]. 3' RACE utilizes a poly(dT) primer to target the poly(A) tail, facilitating the retrieval of 3' terminal sequences [22]. 5' RACE employs various strategies to amplify the 5' end, including terminal transferase tailing, adapter ligation, and the switching mechanism at the 5' end of the RNA template (SMART) approach [22,23]. RACE is a cost-effective and readily accessible technique for most molecular biology laboratories. In a direct comparison study, this method demonstrated higher sensitivity than CaptureSeq in detecting splice junctions [24]. RACE has been combined with tiling arrays or next-generation sequencing (NGS) to resolve complex transcriptional patterns. Kapranov et al. integrated RACE with high-density tiling arrays, revealing the extensive complexity of the human transcriptome, including transcript fusion and interlacing structures [5]. Lagarde et al. developed RACE-Seq, performing 5' and 3' RACE on 398 known long noncoding RNA (lncRNA) exons, followed by high-throughput sequencing using the Roche 454 FLX+ NGS platform, yielding reads with an average length of approximately 600 base pairs (bp) [24]. In our recent studies, RACE was integrated with Nanopore long-read sequencing to determine the complete structure of novel intragenic or intergenic transcripts, using GENSCAN-predicted exons as anchors [25,26]
The inherent complexity of the transcriptome, characterized by dynamic splicing patterns and structural diversity, poses significant analytical challenges. Conventional RACE coupled with Sanger sequencing, while capable of generating relatively long reads, suffers from low throughput and labor-intensive workflows. Conversely, short-read NGS technologies have intrinsic limitations in adequately resolving complex splicing patterns. Although long-read sequencing overcomes read length limitations and enables full-length transcript detection [27], its modest throughput remains a bottleneck for capturing the full diversity of low-abundance transcripts. Therefore, combining targeted RNA enrichment techniques with long-read sequencing offers an effective strategy, providing a practical and streamlined solution for targeted analysis of complex loci. Among long-read sequencing techniques, Nanopore sequencing achieves significantly longer read lengths than PacBio and is more cost-effective, providing unprecedented capability for de novo gene annotation and structural variant detection [19,28,29].
Here, we introduce RACE coupled with Nanopore sequencing (RACE-Nano-Seq), a method designed to efficiently capture full-length transcripts. This approach leverages target locus sequences (annotated exons, predicted exons, or other sequences) as anchors for full-length transcript enrichment via 5'/3' RACE. The enriched cDNA products are then analyzed with Nanopore sequencing and aligned to the corresponding reference genome to enable sensitive transcriptome characterization. This approach is particularly well-suited for detecting low-abundance transcripts, identifying novel exons, and characterizing splicing patterns at specific gene loci. In this protocol, we detail the experimental procedures and analytical pipelines for implementing RACE-Nano-Seq. Additionally, we provide an example demonstrating its application in profiling the transcriptome diversity at a specific gene locus.
Materials and reagents
Biological materials
1. K562 (Cell Bank of Chinese Academy of Sciences, catalog number: TCHu191)
Reagents
1. TRNzol universal reagent (Tiangen, catalog number: DP424)
2. Chloroform (Guoyao, catalog number: 10006818)
3. E.Z.N.A.® Total RNA kit (OMEGA, catalog number: R6834-02). Kit components used in this protocol: HiBind® RNA Mini column, collection tube, RNA wash buffer I, RNA wash buffer II
4. Library preparation VAHTSTM mRNA capture beads (Vazyme, catalog number: N401-02)
5. VAHTS DNA clean beads (Vazyme, catalog number: N411)
6. UltraPureTM DNase/RNase-free distilled water (Invitrogen, catalog number: 10977035)
7. Ethanol (Guoyao, catalog number: 10009218)
8. PrimeScriptTM II 1st Strand cDNA Synthesis kit (Takara, catalog number: 6210A). Kit components used in this protocol: 10 mM dNTP mix, 5× PrimeScript II buffer, 200 U/μL PrimeScript II reverse transcriptase, 40 U/μL RNase inhibitor
9. Terminal transferase (NEB, catalog number: M0315): 20 U/μL terminal transferase, 10× terminal transferase buffer, 10× CoCl2
10. PrimeSTAR® GXL DNA polymerase (Takara, catalog number: R050A). Kit components used in this protocol: 5× PrimeSTAR GXL buffer, PrimeSTAR GXL DNA polymerase (1.25 U/μL), dNTP mix (2.5 mM each)
11. Agarose (Invitrogen, catalog number: 75510-019)
12. 50× TAE buffer (Solarbio, catalog number: T1060)
13. 10,000× SuperRed (Biosharp, catalog number: BS354A)
14. Ligation Sequencing kit (Oxford Nanopore Technologies, catalog number: SQK-LSK114). Kit components used in this protocol: AMPure XP beads, ligation adapters, ligation buffer, flow cell tether, flow cell flush, sequencing buffer, library beads, short fragment buffer, elution buffer
15. NEBNext FFPE Repair Mix (NEB, catalog number: M6630), includes: NEBNext FFPE DNA repair mix, NEBNext FFPE DNA repair buffer
16. NEBNext Ultra II End Repair/dA-tailing module (NEB, catalog number: E7546), includes: ultra II end-prep reaction mix, ultra II end-prep enzyme buffer
17. NEBNext Quick Ligation module (NEB, catalog number: E6056), includes NEBNext Quick T4 DNA ligase
18. Qubit dsDNA HS Assay kit (ThermoFisher, catalog number: Q32851)
19. InvitrogenTM UltraPureTM BSA (Invitrogen, catalog number: AM2616)
20. Equalbit 1× dsDNA HS Assay kit (Vazyme, catalog number: EQ121-01)
Solutions
1. 70% ethanol (see Recipes)
2. 80% ethanol (see Recipes)
3. 1× TAE buffer (see Recipes)
4. 1% agarose gel (see Recipes)
Recipes
1. 70% ethanol
| Reagent | Final concentration | Volume |
|---|---|---|
| Ethanol | 70% (v/v) | 700 μL |
| UltraPureTM DNase/RNase-free distilled water | 30% (v/v) | 300 μL |
Note: Prepare fresh 70% and 80% ethanol solutions immediately before use and adjust the volume based on reaction needs.
2. 80% ethanol
| Reagent | Final concentration | Volume |
|---|---|---|
| Ethanol | 80% (v/v) | 800 μL |
| UltraPureTM DNase/RNase-free distilled water | 20% (v/v) | 200 μL |
3. 1× TAE buffer
| Reagent | Final concentration | Volume |
|---|---|---|
| 50× TAE buffer | 1× | 1 mL |
| Ultrapure water (lab-purified) | n/a | 49 mL |
4. 1% agarose gel
| Reagent | Final concentration | Quantity/volume |
|---|---|---|
| Agarose | 1% (w/v) | 0.3 g |
| 10,000× SuperRed | 1× | 3 μL |
| 1× TAE buffer | n/a | 30 mL |
Laboratory supplies
1. 1.5 mL EP tubes (Axygen, catalog number: MCT-150-C)
2. 200 μL PCR tubes (Axygen, catalog number: PCR-02-C)
3. 10 μL pipette tips (Axygen, catalog number: TF-300)
4. 20 μL pipette tips (Axygen, catalog number: TF-20)
5. 200 μL pipette tips (KIRGEN, catalog number: KG5213-L)
6. 1,000 μL pipette tips (KIRGEN, catalog number: KG5313-L)
Equipment
1. Spectrophotometer (Merinton, model: SMA6000)
2. Fluorescence imaging system (Tanon, model: Tanon 3500R)
3. Qubit 4 fluorometer (Thermo Fisher, catalog number: Q33238)
4. C1000 TouchTM thermal cycler (Bio-Rad, model: C1000)
5. Oxford Nanopore PromethION (Oxford Nanopore Technologies)
6. FLO-PRO002 R10.4 flow cell (Oxford Nanopore Technologies)
7. Rotator mixer (Qilinbeier, model: BE-1100)
8. Magnetic rack (Promega, catalog number: Z5332)
9. Direct-Pure UP Ultrapure & RO Lab Water System (Rephile)
Software and datasets
1. Guppy (v4.3.6)
2. NanoFilt (2.8.0, https://github.com/wdecoster/nanofilt/)
3. Minimap2 (v2.17-r941, https://github.com/lh3/minimap2/)
4. Samtools (1.10, https://github.com/samtools/samtools/)
5. BEDTools (v2.30.0, https://bedtools.readthedocs.io/en/latest/)
6. GRCh38/hg38 (https://hgdownload.cse.ucsc.edu/goldenpath/hg38/bigZips/)
7. Bedparse (v0.2.3, https://github.com/tleonardi/bedparse/tree/b2833706a006504b267b9a0692334a7d18e44e5c/)
8. Encyclopedia of DNA Elements (ENCODE) Candidate Cis-Regulatory Elements (https://genome.ucsc.edu/)
9. ENCODE H3K4Me3 Mark in K562 cell line (https://genome.ucsc.edu/)
10. Functional Annotation of the Mammalian Genome 5 (FANTOM5) CAGE (https://fantom.gsc.riken.jp/5/datafiles/reprocessed/hg38_latest/basic/)
Procedure
文章信息
稿件历史记录
提交日期: Mar 31, 2025
接收日期: Jun 3, 2025
在线发布日期: Jun 19, 2025
出版日期: Jul 5, 2025
版权信息
© 2025 The Author(s); This is an open access article under the CC BY-NC license (https://creativecommons.org/licenses/by-nc/4.0/).
如何引用
Tang, L., Xu, D. and Kapranov, P. (2025). RACE-Nano-Seq: Profiling Transcriptome Diversity of a Genomic Locus. Bio-protocol 15(13): e5374. DOI: 10.21769/BioProtoc.5374.
分类
生物信息学与计算生物学
系统生物学 > 转录组学 > RNA测序
分子生物学 > RNA > RNA 测序
您对这篇实验方法有问题吗?
在此处发布您的问题,我们将邀请本文作者来回答。同时,我们会将您的问题发布到Bio-protocol Exchange,以便寻求社区成员的帮助。