

Protein Structural Characterization Using Electron Transfer Dissociation and Hydrogen Exchange-Mass Spectrometry
结合电子转移解离与氢交换质谱的蛋白质结构特征分析方法
发布: 2025年06月20日第15卷第12期 DOI: 10.21769/BioProtoc.5350 浏览次数: 1992
评审: Dhananjay D ShindeAnonymous reviewer(s)
参见作者原研究论文
The authors used this protocol in:

Nov 2021
Advertisement










Abstract
Intermediate states are often populated during the folding and unfolding reactions of a protein, and their detection is very challenging as they form transiently. Structural characterization of these short-lived intermediate species is difficult as it requires high-resolution methodologies. Hydrogen exchange-mass spectrometry (HX-MS) can identify and yield direct structural information on folding and unfolding intermediates, as well as information about the cooperativity of the folding or unfolding processes. The mass distributions of intact protein molecules are obtained first to determine their exchange pattern. Then, segment-specific structural information is obtained by analyzing the fragments of the protein. Enzymatic digestion is widely used with HX to determine the sequence-specific structural changes that occur to the protein during folding or unfolding. However, if a protein is an inhibitor of the protease, then alternative methodologies are required. Using electron transfer dissociation (ETD), it is possible to fragment the protein inside a mass spectrometer, and segment-specific structural changes occurring during the folding and unfolding process can be determined. In the case of HX-ETD-MS, protein molecules are first allowed to undergo HX, followed by their fragmentation. Deuterium retention in each fragment is measured. Very little, if any, scrambling of deuterium across fragments occurs during ETD-enabled fragmentation; hence, there is little scope for misinterpretation of the HX data.
Key features
• Analysis of intact protein data allows the identification of the intermediate states even in native and native-like conditions.
• Precursor mass is determined from the intact protein analysis.
• Fragments undergoing cooperative and non-cooperative transitions during protein unfolding can be distinguished from each other using HX-ETD-MS.
• Analysis of the protein fragments that are obtained using ETD also enables the determination of the sequence of structural changes occurring in the protein.
• HX-ETD-MS can provide structural insights into transiently formed intermediate states.
Keywords: Non-cooperative (非协同作用)Graphical overview
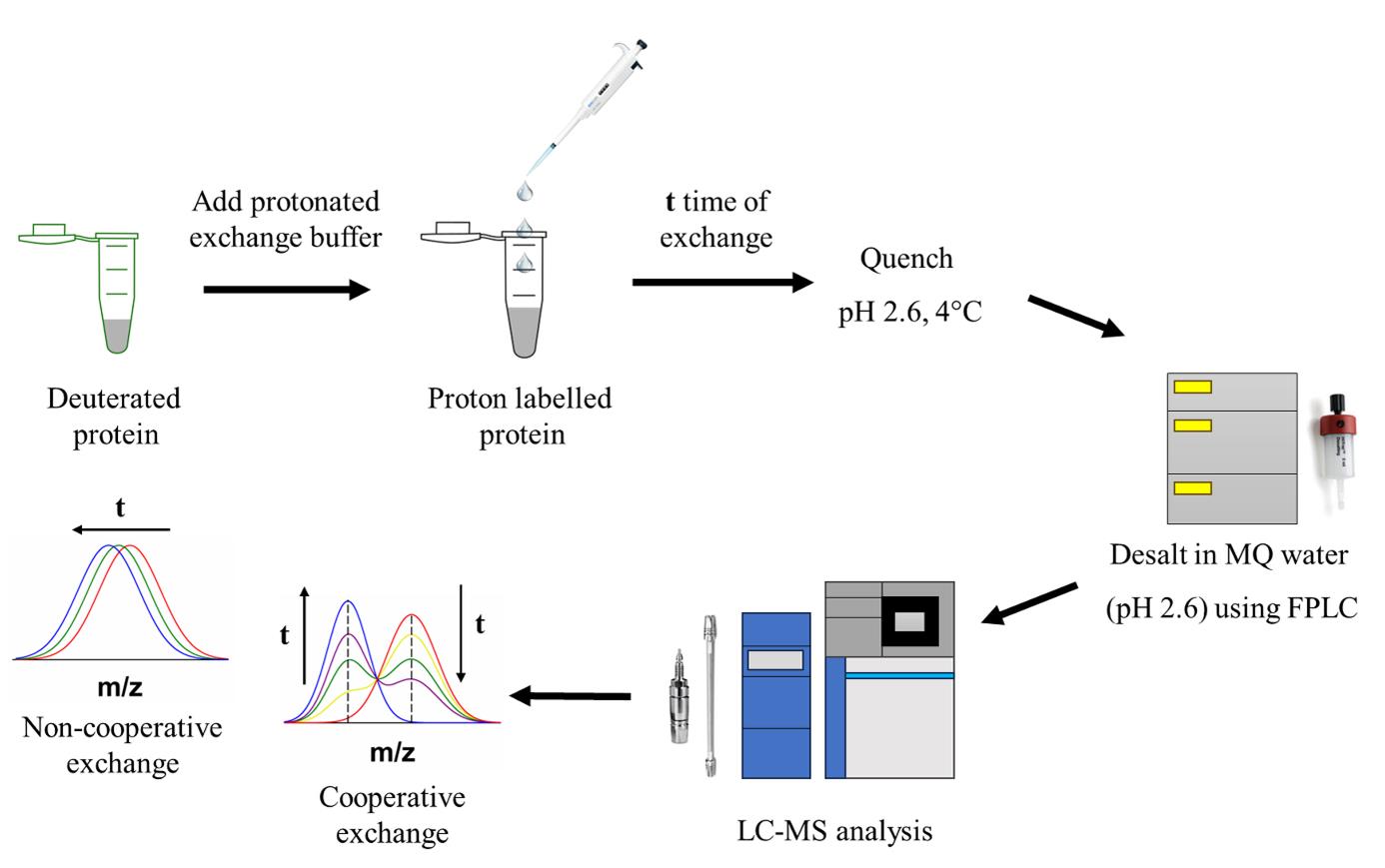
Protein structural characterization using hydrogen exchange–electron transfer dissociation–mass spectrometry (HX-ETD-MS)
Background
Protein folding or unfolding reactions can vary from being highly cooperative to being non-cooperative in nature. A cooperative transition does not involve the formation of any intermediate states [1,2], whereas multiple intermediates are populated during a non-cooperative transition [3–7]. If the partially unfolded intermediate states are not populated to detectable extents, then the folding or unfolding reaction might appear to be a cooperative process [8–11]. Conventional ensemble-averaging probes such as fluorescence or circular dichroism (CD), which are commonly used to study protein folding and unfolding reactions, are unable to detect sparsely populated intermediate species. High-resolution methodologies are required that can not only detect co-existing conformations but also quantify them. Methodologies such as hydrogen exchange (HX) [12–16], sulfhydryl exchange (SX) [17,18], time-resolved fluorescence resonance energy transfer (trFRET) [19,20], and single-molecule FRET (smFRET) [21] have the potential to quantitatively measure the populations of co-existing conformations, and hence have been used to delineate the sequence of structural changes that occur during folding and unfolding. These methodologies have also enabled the structural characterization of transiently formed intermediate states very efficiently [22–25].
However, fluorescence studies require labeling of the protein with a high quantum yield dye, which is not very straightforward. Incorporation of the dye might also have some implications for the secondary and tertiary structure of the protein. On the other hand, HX studies typically rely on enzymatic digestion by pepsin for fragmenting the protein in order to determine where the protein exchange occurred. While pepsin digestion is easy to carry out [12,23,26,27], some proteins are inhibitors of pepsin, disallowing its use [16,28]. As electron transfer dissociation (ETD) involves the fragmentation of the protein inside the mass spectrometer, it has the advantage that it can be used for all proteins. This method was discovered in 2004 [29]. Briefly, a small chemical molecule having a lower electron affinity is allowed to transfer its electron to the protein backbone under a set of conditions. This energizes the protein molecule, leading to its fragmentation at the N-Cα bond, generating c and z ions. ETD has been used in the past to study posttranslational modifications of protein [30,31] and protein–protein interactions [32]. Coupling ETD with HX allows the study of the structural changes occurring in a protein during folding, and the structural characterization of the intermediate states [7,16,28]. The disadvantage of this method is the very low fragmentation efficiency of the protein. The intensities of the peptides are often very low, and many peptides cannot be detected reliably.
HX-MS is not only used to study protein folding but has also been used to study protein–protein interactions, protein–drug interactions, etc. Moreover, mass spectrometry alone has a broad application in proteomics studies. For larger proteins, enzymatic digestion does not give full coverage of the sequence; hence, enzymatic digestion can be coupled with ETD to increase the sequence coverage. This will enable the reliable detection and identification of the proteins and their interacting partners [33].
Materials and reagents
Reagents
1. Guanidine hydrochloride (GdnHCl) (United States Biochemicals, catalog number: 75823)
2. Deuterium oxide (D2O) (Sigma-Aldrich, catalog number: 151882)
3. Tris base (Sigma-Aldrich, catalog number: T1378)
4. Glycine (United States Biochemicals, catalog number: 75826)
5. Sodium hydroxide (Fischer chemicals, catalog number: S318)
6. Hydrochloric acid (HCl) (Fischer chemicals, catalog number: A144S)
7. Sodium deuteroxide (NaOD) (Sigma-Aldrich, catalog number: 176788)
8. Deuterium chloride (DCl) (Sigma-Aldrich, catalog number: 543047)
9. 1,4-dicyano benzene (Sigma-Aldrich, catalog number: D76722)
10. Formic acid (Merck, catalog number: 5.33002.0050)
11. Acetonitrile (Merck, catalog number: 1.00029.2500)
12. Horse heart myoglobin (Sigma-Aldrich, catalog number: M1882)
13. Glu-fibrinopeptide (Waters, catalog number: 700004729)
14. Ice-cold Mili-Q water at pH 2.6 (adjust pH with LC–MS-grade formic acid)
Solutions
1. Labeling buffer (see Recipes)
2. Quenching buffer (see Recipes)
3. Tris D2O buffer (see Recipes)
Recipes
1. Labeling buffer (50 mL)
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| Tris (2 M) | 20 mM | 500 μL |
| H2O | n/a | see note* |
| Total | n/a | 50 mL |
*Add water up to 45 mL, adjust pH to 8 by adding HCl, and make up the volume to 50 mL. Filter the solution using a 0.22 μm filter.
2. Quenching buffer (50 mL)
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| Glycine (2 M) | 100 mM | 2.5 mL |
| Guanidine hydrochloride | 8 M | 38.2 g |
| H2O | n/a | see note* |
| Total | n/a | 50 mL |
*Add water up to 45 mL. While dissolving GdnHCl, put the beaker in hot water to facilitate the hydration of GdnHCl. Adjust the pH to 2.2 by adding HCl, then make up the volume to 50 mL. Filter the solution using a 0.22 μm filter.
3. Tris D2O buffer (10 mL)
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| Tris in D2O (2 M) | 10 mM | 50 μL |
| D2O | n/a | see note* |
| Total | n/a | 10 mL |
*Add D2O up to 8 mL and adjust pH to 8.2 by adding DCl. Then, make up the volume to 10 mL and filter the solution using a 0.22 μm filter.
Laboratory supplies
1. 1.5 mL microcentrifuge tube (Tarsons, catalog number: 500010)
2. 0.22 µm filters (Sartorius, catalog number: 16534K)
3. CD cuvette (Starna Scientific, catalog number: 21/Q/1)
4. Hamilton glass syringe (Hamilton company, catalog number: 81165/00)
5. Sephadex G-25 Hi-trap desalting column (Cytiva, catalog number: 17140801)
6. C18 VanGuard trap column (Waters, catalog number: 186003975)
7. C18 analytical column (Waters, catalog number: 186002346)
Equipment
1. High-definition mass spectrometer (Waters Corporation, model: Synapt G2)
2. UPLC (Waters corporation, model: nanoAcquity)
3. HDX module (Waters corporation)
4. Chromatographic system (Postnova, model: AF4)
5. Cary spectrophotometer (Agilent)
6. Refractometer (Abbe)
7. pH meter (Thermo Scientific)
8. CD spectrophotometer (Jasco J15)
Software and datasets
1. MassLynx (SCN 781, Waters)
2. Microsoft Excel (MS office home and student 2016) from Microsoft
3. SigmaPlot (v12.0, Systat Software Inc.)
4. OriginPro (v9.0, OriginLab)
5. Pymol molecular graphics system (v2.3.3)
Procedure
文章信息
稿件历史记录
提交日期: Jan 6, 2025
接收日期: May 15, 2025
在线发布日期: Jun 5, 2025
出版日期: Jun 20, 2025
版权信息
© 2025 The Author(s); This is an open access article under the CC BY-NC license (https://creativecommons.org/licenses/by-nc/4.0/).
如何引用
Bhattacharjee, R. and Udgaonkar, J. B. (2025). Protein Structural Characterization Using Electron Transfer Dissociation and Hydrogen Exchange-Mass Spectrometry. Bio-protocol 15(12): e5350. DOI: 10.21769/BioProtoc.5350.
分类
生物化学 > 蛋白质 > 结构
您对这篇实验方法有问题吗?
在此处发布您的问题,我们将邀请本文作者来回答。同时,我们会将您的问题发布到Bio-protocol Exchange,以便寻求社区成员的帮助。