

Chloroform/Methanol Protein Extraction and In-solution Trypsin Digestion Protocol for Bottom-up Proteomics Analysis
用于自下而上蛋白质组学分析的氯仿/甲醇蛋白提取与溶液内胰蛋白酶消化方法
发布: 2024年08月20日第14卷第16期 DOI: 10.21769/BioProtoc.5055 浏览次数: 3122
评审: Dennis ProvinceNeha NandwaniAnonymous reviewer(s)
参见作者原研究论文
The authors used this protocol in:

Jan 2024
Advertisement









Abstract
Bottom-up proteomics utilizes sample preparation techniques to enzymatically digest proteins, thereby generating identifiable and quantifiable peptides. Proteomics integrates with other omics methodologies, such as genomics and transcriptomics, to elucidate biomarkers associated with diseases and responses to drug or biologics treatment. The methodologies employed for preparing proteomic samples for mass spectrometry analysis exhibit variability across several factors, including the composition of lysis buffer detergents, homogenization techniques, protein extraction and precipitation methodologies, alkylation strategies, and the selection of digestion enzymes. The general workflow for bottom-up proteomics consists of sample preparation, mass spectrometric data acquisition (LC-MS/MS analysis), and subsequent downstream data analysis including protein quantification and differential expression analysis. Sample preparation poses a persistent challenge due to issues such as low reproducibility and inherent procedure complexities. Herein, we have developed a validated chloroform/methanol sample preparation protocol to obtain reproducible peptide mixtures from both rodent tissue and human cell line samples for bottom-up proteomics analysis. The protocol we established may facilitate the standardization of bottom-up proteomics workflows, thereby enhancing the acquisition of reliable biologically and/or clinically relevant proteomic data.
Key features
• Tissue/cell pellet sample preparation for bottom-up proteomics
• Chloroform/methanol protein extraction from murine tissue samples
• In-solution trypsin digestion proteomics workflow
Keywords: Proteomics (蛋白质组学)Graphical overview
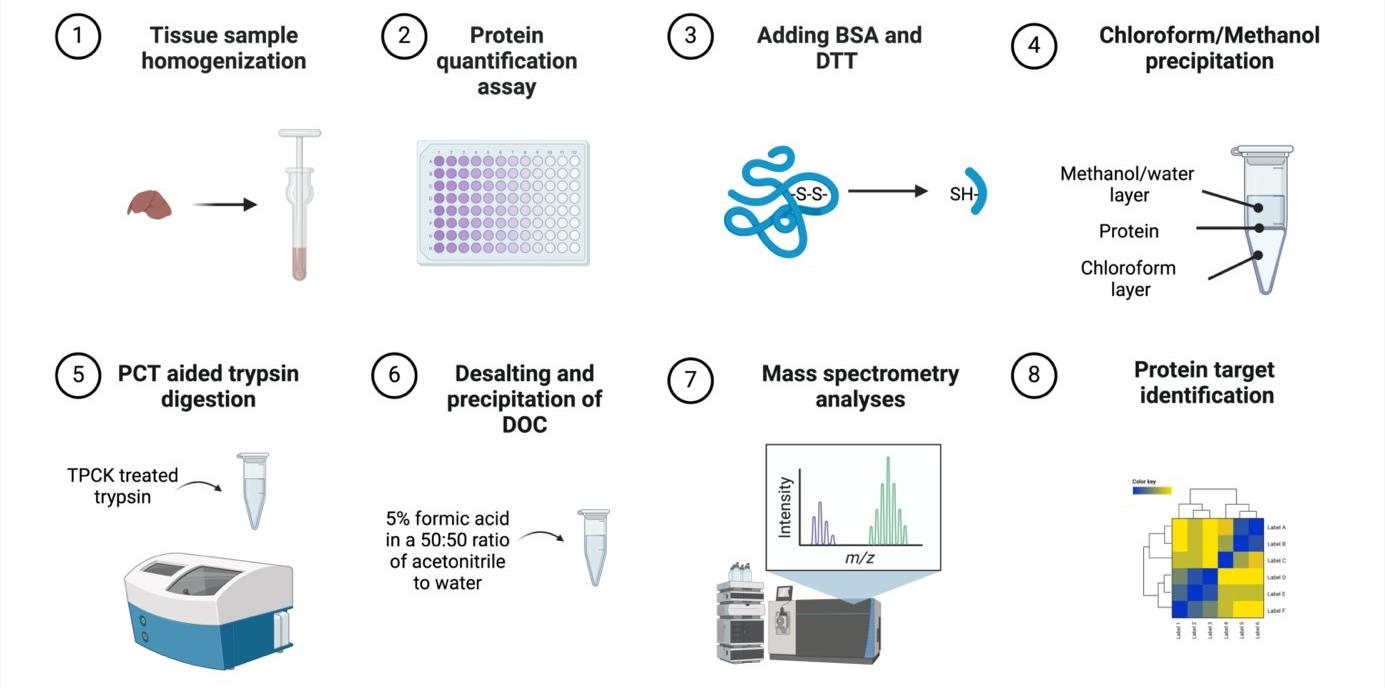
Murine tissue sample chloroform/methanol protein extraction workflow for bottom-up proteomics analyses
Background
Sample preparation is a critical component of bottom-up proteomics analyses, yet it is often hindered by time constraints, performer errors, and intensive processes. Further, there remains a scientific need to develop reproducible, precise, and successful sample preparation protocols to advance proteomic discovery [1]. There are numerous sample preparation methods due to differences in sample type, protein heterogeneity, and physicochemical properties, which creates difficulty in standardizing workflows [2,3]. In-solution digestion (ISD) primarily employs varied buffers such as denaturants and detergents to denature protein samples followed by protein precipitation with organic solvents, such as chloroform/methanol, alcohol, or acetone [2]. A recent novel ISD protocol, sample preparation by easy extraction and digestion (SPEED), denatures proteins without the use of denaturants and detergents but rather through the use of trifluoracetic acid [2]. In-gel digestion (IGD), in contrast to ISD, involves the separation of proteins by size via gel electrophoresis, which has the capacity to reduce contaminants [4]. IGD is often coupled with SDS-PAGE, which utilizes the strong detergent SDS [5,6]. SDS may decrease the efficacy of proteases, such as trypsin, as well as reduce the signal/noise ratio, thereby limiting proteome coverage [5]. Therefore, it is advantageous to avoid the use of SDS in sample digestion steps or buffer solutions to prevent adverse effects and SDS removal steps. Similarly, another sample preparation method popularly employed includes device-based approaches such as filter-aided sample preparation (FASP) where SDS is also added to protein samples [7]. Samples are then filtered, and the protein is digested to elute peptides. However, FASP sample preparation protocols are time-consuming and have resulted in considerable variability, leading to the development of other methods [7]. Recently, the suspension trapping (S-Trap) method has garnered attention due to its protein extraction method via denatured size, which has improved time constraints and complexities in sample preparation [8]. However, the S-Trap method typically uses 5% SDS as a lysis buffer, and the inherent disadvantages of SDS still remain [7].
Therefore, ISD remains a promising sample preparation method compared to other approaches, yet standardization of methods for animal and cell culture samples is warranted. Herein, we developed an ISD protocol consisting of denaturation, alkylation, chloroform/methanol protein extraction, and digestion steps, as ISD methods are commonly employed in sample preparation of animal tissue samples [9]. Specifically, ISD complemented by a chloroform/methanol protein extraction method may be a reliable technique for animal and human tissue samples bottom-up proteomics as it is a fairly straightforward and fast method, which prevents protein degradation [10]. Chloroform/methanol protein extraction is a quantitative method first developed in 1984, utilizing a phase separation to precipitate proteins in solution; it has since been shown to be advantageous over acetone and ethanol for total peptides and peptides without missed cleavages [2,11]. Further, chloroform/methanol protein extraction produces a protein pellet that does not contain contaminants without reducing protein quantities [11]. In comparison to IGD, ISD is less error-prone and costly and has a higher likelihood of high peptide yield [2,4]. Further, studies have found that ISD is advantageous to identify peptides from the whole proteome, while other methods such as FASP may be better suited for specific protein recovery such as membrane proteins [12]. Our bottom-up proteomics tissue sample preparation technique using classic ISD and chloroform/methanol protein extraction methods provides a valid and reproducible workflow with protein yields suitable for in-depth, whole-proteome analyses.
Materials and reagents
Biological materials
Frozen mouse tissue sample
Note: This protocol has also been successfully conducted using cell lysis samples in the following manuscripts:
Li et al. (2024). Anti-Ferroptotic Effect of Cannabidiol in Human Skin Keratinocytes Characterized by Data-Independent Acquisition-Based Proteomics. J Nat Prod. DOI: 10.1021/acs.jnatprod.3c00759. (Figures 3 and 4) [13].
Li et al. (2023). Cannflavins A and B with Anti-Ferroptosis, Anti-Glycation, and Antioxidant Activities Protect Human Keratinocytes in a Cell Death Model with Erastin and Reactive Carbonyl Species. Nutrients. DOI: 10.3390/nu15214565. (Figure 2) [14].
Dry ice (Airgas)
Urea (Sigma-Aldrich, catalog number: 51457-500 mL)
Triethylammonium bicarbonate buffer (TEAB) (Sigma-Aldrich, catalog number: T7408-100 mL)
PierceTM BCA Protein Assay kit with dilution-freeTM BSA protein standards (Thermo Fisher Scientific, catalog number: A55865)
Bovine serum albumin (BSA) (Sigma-Aldrich, catalog number: A9418-5G)
DL-Dithioreitol (DTT) (Sigma-Aldrich, catalog number: D9779-1G)
Iodoacetamide (IAA) (Sigma-Aldrich, catalog number: I1149-5G)
Ammonium bicarbonate (Sigma-Aldrich, catalog number: A6141-25G)
Sodium deoxycholate (DOC) (Sigma-Aldrich, catalog number: 30970-25G)
Trypsin w/ CaCl2 (TPCK-treated), 10 pack (SCIEX, catalog number: 4445250)
Methanol CHROMASOLV LC-MS (Honeywell, catalog number: 34966-4L)
Chloroform-isoamyl alcohol mixture (Sigma-Aldrich, catalog number: 25666-100ML)
Milli-Q water
Formic acid (Sigma-Aldrich, catalog number: 695076-100ML)
Acetonitrile (ACN) CHROMASOLV LC-MS (Honeywell, catalog number: 34967-4X4L)
TEAB (50 mM) and urea (8M) homogenization buffer (see Recipes)
50 mM ammonium bicarbonate (pH ~8) containing 3% w/v sodium deoxycholate (DOC) (see Recipes)
5% formic acid in acetonitrile and water (50:50 ratio) (see Recipes)
TEAB (50 mM) and urea (8M) homogenization buffer
Reagent Final concentration TEAB 50 mM Urea 8 M 50 mM ammonium bicarbonate (pH ~8) containing 3% w/v sodium deoxycholate (DOC)
Reagent Final concentration Ammonium bicarbonate 50 mM DOC 3% 5% formic acid in acetonitrile and water (50:50 ratio)
Reagent Final concentration Acetonitrile 47.5% Water 47.5% Formic acid 5%
Laboratory supplies
Pipette Tips (TipOne 10, 200, 1,000 μL) (USA Scientific, catalog numbers: 1111-3800, 1110-1800, 1111-2821)
Pipettes (ErgoOne Single-Channel Pipette 2.5, 10, 200, 1,000 μL) (USA Scientific, catalog numbers: 7100-0125, 7100-0510, 7100-2200, 7110-1000)
Soft tissue homogenizing mix 1.4 mm ceramic (2 mL tubes) nuclease free (Omni International, catalog number: 19-627)
1 mL Dounce tissue grinder (Avantor, catalog number: 357538)
Pour boat weigh dish 2-1/4"l × 1-3/4"w × 5/16"d, 20 mL cap (Wilkem Scientific, catalog number: 10177901)
96-well tissue culture plates (Cell Treat, catalog number: 229197)
96 150 μL PCT microtubes (Pressure Biosciences, Inc., catalog number: MTWS-MT-RK)
150 μL PCT microcaps (Pressure Biosciences, Inc., catalog number: MTWS-MC150-RK)
Pipet tip gel loading .57 mm O.D. 200 μL round non-sterile (Wilkem Scientific, catalog number: LABB13790)
Microcentrifuge tube 0.5 mL non-sterile (Cell Treat, Wilkem Scientific, catalog number: 72316004)
Microcentrifuge tube 1.5 mL non-sterile (Cell Treat, Wilkem Scientific, catalog number: 229441)
PK100 amber glass certified S/T vial (Sigma-Aldrich, catalog number: 29386-U)
Advantage 150 μL volume, 5 × 30 mm bottom spring glass inserts (Analytical Sales and Services Inc., catalog number: 20501)
Equipment
Bead Ruptor elite bead mill homogenizer (Omni International, model: 19-042E)
Microplate reader SpectraMax M2 (Molecular Devices, model: MDM2)
Pressure cycling technologies Barocycler (Pressure Biosciences Inc., model: NEP2320)
Circulating heater water bath (Thermo Fisher Scientific, model: CH 100)
Microcap tool Series 3 (Pressure Biosciences Inc., model: MTWS-CR-03)
Microtube adapter kit (Pressure Biosciences Inc., catalog number: MTTB-KEXT)
Centrifuge 5810 R (Eppendorf, model: 5811F)
Reciprocal shaking water bath (Precision Scientific, model: 66800)
Fisher vortex Genie 2TM (Fisher Scientific, catalog number: 12-812)
Software and datasets
Prism v10.0.3 (GraphPad, 07/05/2024)
Procedure
文章信息
稿件历史记录
提交日期: May 20, 2024
接收日期: Jul 17, 2024
在线发布日期: Aug 2, 2024
出版日期: Aug 20, 2024
版权信息
© 2024 The Author(s); This is an open access article under the CC BY license (https://creativecommons.org/licenses/by/4.0/).
如何引用
Puopolo, T., Seeram, N. P. and Liu, C. (2024). Chloroform/Methanol Protein Extraction and In-solution Trypsin Digestion Protocol for Bottom-up Proteomics Analysis. Bio-protocol 14(16): e5055. DOI: 10.21769/BioProtoc.5055.
分类
系统生物学 > 蛋白质组学 > 整个机体
生物化学 > 蛋白质 > 分离和纯化
分子生物学 > 蛋白质 > 检测
您对这篇实验方法有问题吗?
在此处发布您的问题,我们将邀请本文作者来回答。同时,我们会将您的问题发布到Bio-protocol Exchange,以便寻求社区成员的帮助。