

Phylogenetic Inference of Homologous/Orthologous Genes among Distantly Related Plants
远缘植物中同源/直系同源基因的系统发育推断

发布: 2023年12月05日第13卷第23期 DOI: 10.21769/BioProtoc.4893 浏览次数: 2519
评审: Xin QiaoYao XiaoYe XuAnonymous reviewer(s)
参见作者原研究论文
The authors used this protocol in:

Apr 2023
Advertisement









Abstract
The recent surge in plant genomic and transcriptomic data has laid a foundation for reconstructing evolutionary scenarios and inferring potential functions of key genes related to plants’ development and stress responses. The classical scheme for identifying homologous genes is sequence similarity–based searching, under the crucial assumption that homologous sequences are more similar to each other than they are to any other non-homologous sequences. Advances in plant phylogenomics and computational algorithms have enabled us to systemically identify homologs/orthologs and reconstruct their evolutionary histories among distantly related lineages. Here, we present a comprehensive pipeline for homologous sequences identification, phylogenetic relationship inference, and potential functional profiling of genes in plants.
Key features
• Identification of orthologs using large-scale genomic and transcriptomic data.
• This protocol is generalized for analyzing the evolution of plant genes.
Keywords: Homolog (同源物)Background
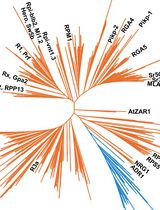
Evolution of plant genes is inextricably coupled with various evolutionary events, including endosymbiotic events, whole-genome duplication/triplication (WGD/T), gene loss, and horizontal gene transfer (Zhang et al., 2022). Archaeplastida, including green plants (Viridiplantae), glaucophytes (Glaucophyta), and red algae (Rhodophyta), originate anciently and most of them have experienced multiple WGD/T events, resulting in dramatic changes in copy numbers and complicated evolutionary trajectories of their homologous genes (Qiao et al., 2019). Homologs, orthologs, and paralogs are important concepts for the evolutionary classification of genes, being prevalent in recent comparative genomic studies. Homologs are genes sharing a common origin; orthologs and paralogs are two types of homologous genes, which separately evolved via speciation and gene duplication (Thornton and DeSalle, 2000; Koonin, 2005). Homologous genes generally have a relatively higher degree of sequence similarity than non-homologous genes. Sequence similarity–based searching and phylogenetic analyses are useful tools for identifying homologous sequences of genes and reconstructing their evolutionary routes.
Although the definition of homology/orthology has nothing to do with biological functions, there are major functional connotations (Koonin, 2005). Homologous/orthologous genes among different plants typically perform similar or equivalent functions, which is theoretically plausible and empirically supported. Thus, for a newly identified gene in non-model plants, identifying its homologs/orthologs in model plants or crops that have well-documented functional annotations is very useful to assign its possible functions. Phylogenetic analyses can reconstruct the evolutionary trajectories of homologs/orthologs among various species, which can facilitate the understanding of the molecular mechanisms underpinning its biological functions. Here, taking the acetyltransferase like protein HOOKLESS1 (HLS1) as an example (Lehman et al., 1996; Li et al., 2004), we provide a detailed procedure for homologs/orthologs identification using large-scale genomic and transcriptomic data of distantly related plants. This protocol includes generalized steps and parameters for evolutionary analyses of plant genes, and some of these steps and parameters can be customized based on the genes of interest.
Equipment
Server with a 64-bit Linux-based operating system (Ubuntu 18.04.6 LTS): 512 GB RAM and Intel Xeon (R) Gold 6238 CPU
Desktop with a Windows 10 operating system: Intel Core i5-8300H CPU and 8 GB RAM
Software and datasets
Software and databases used in this protocol are as follows:
Miniconda3-py39_4.12.0-Linux-x86_64 (https://mirrors.tuna.tsinghua.edu.cn/anaconda/miniconda/Miniconda3-py39_4.12.0-Linux-x86_64.sh)
TBtools v1.120 (Chen et al., 2020)
Diamond v2.1.7.161 (Buchfink et al., 2015)
MAFFT v7.453 (Katoh and Standley, 2013)
trimAL v1.4.rev15 (Capella-Gutiérrez et al., 2009)
IQ-TREE v2.2.2.6 (Minh et al., 2020)
InterProScan 5.63-95.0 (Jones et al., 2014)
1KP dataset (One Thousand Plant Transcriptomes Initiative, 2019)
MEME 5.5.3 (Bailey and Elkan, 1994)
iTOL (Interactive Tree Of Life) (Letunic and Bork, 2021)
Jalview v2.11.2.0 (Waterhouse et al., 2009)
Procedure
文章信息
版权信息
© 2023 The Author(s); This is an open access article under the CC BY-NC license (https://creativecommons.org/licenses/by-nc/4.0/).
如何引用
Xu, Z., Sun, W., Zhu, Z., Zhong, B. and Zhang, Z. (2023). Phylogenetic Inference of Homologous/Orthologous Genes among Distantly Related Plants. Bio-protocol 13(23): e4893. DOI: 10.21769/BioProtoc.4893.
分类
系统生物学 > 基因组学 > 种系遗传学
生物信息学与计算生物学
您对这篇实验方法有问题吗?
在此处发布您的问题,我们将邀请本文作者来回答。同时,我们会将您的问题发布到Bio-protocol Exchange,以便寻求社区成员的帮助。