

Single-cell Damagenome Profiling by Linear Copying and Splitting based Whole Genome Amplification (LCS-WGA)
基于线性复制和分裂的全基因组扩增法(LCS-WGA)分析单细胞损伤组
(*contributed equally to this work) 发布: 2022年03月20日第12卷第6期 DOI: 10.21769/BioProtoc.4357 浏览次数: 3587
评审: Gal HaimovichKirsten A. CoprenAnonymous reviewer(s)

参见作者原研究论文
The authors used this protocol in:

Jul 2021
Advertisement








Abstract
Spontaneous DNA damage frequently occurs on the human genome, and it could alter gene expression by inducing mutagenesis or epigenetic changes. Therefore, it is highly desired to profile DNA damage distribution on the human genome and identify the genes that are prone to DNA damage. Here, we present a novel single-cell whole-genome amplification method which employs linear-copying followed by a split-amplification scheme, to efficiently remove amplification errors and achieve accurate detection of DNA damage in individual cells. In comparison to previous methods that measure DNA damage, our method uses a next-generation sequencing platform to detect misincorporated bases derived from spontaneous DNA damage with single-cell resolution.
Keywords: DNA damage (DNA损伤)Background
Spontaneous DNA damage, which is usually caused by exposure to environmental chemicals or the free radicals generated from metabolic processes, frequently occurs in our genome. If the damage is not repaired correctly, it could lead to permanent change in the genomic sequence and sabotage gene functions (Ames et al., 1993; De Bont and van Larebeke, 2004; Tudek et al., 2010; Dizdaroglu et al., 2015). Besides mutagenesis, DNA damage could also perturb the local epigenetic stability and, as a result, alter gene expression patterns (Dabin et al., 2016). The non-uniform distribution of DNA damage on our genome could result from various levels of vulnerability to DNA damage in different genes. With high-damage genes being the potential Achilles’ heels of our genome, the alteration in expression of these genes could directly contribute to the development of human multifactorial diseases. Therefore, the accurate examination of the distribution of DNA damage and identification of high-damage genes are greatly desirable.
So far, high-performance liquid chromatography (HPLC)-based methods, gas chromatography/mass spectrometry (GC/MS)-based methods, and chromatin immunoprecipitation (ChIP)-based methods have been the primary approaches in measuring DNA damage. HPLC and GC/MS based methods are mainly used to measure the global levels of various DNA modifications (De Bont and van Larebeke, 2004; Wagner et al., 1992; Galashevskaya et al., 2013). However, since HPLC and GC/MS-based approaches require the DNA samples to be hydrolyzed into mononucleotides, the sequence context information cannot be recovered. Furthermore, the extensive treatment at the DNA hydrolysis step could introduce a significant amount of artificial DNA damage, which leads to large technical variations. On the other hand, ChIP-based methods could provide a sequence-based measurement of DNA damage by using antibodies to pull down the DNA fragments with damaged DNA (Yoshihara et al., 2014; Garcia-Nieto et al., 2017; Ding et al., 2017; Poetsch et al., 2018; Wu et al., 2018; Amente et al., 2019). However, such methods largely depend on the availability of antibodies that could recognize specific types of DNA damage, and do not have single-base resolution. The artificial damage induced during the DNA shearing step could lead to the overestimation of DNA damage levels.
Recently, we have reported a new single-cell whole-genome amplification (WGA) method, called linear copying and splitting based whole genome amplification (LCS-WGA), which can allow us to achieve genome-wide profiling of DNA damage (Zhu et al., 2021), since DNA damage can induce base misincorporation during amplification (Purmal et al., 1994, 1998; Gehrke et al., 2013). These misincorporated bases can then be directly identified in the sequencing data. We refer to damage-associated single-nucleotide variants as damSNVs. The main advantage of the single-cell WGA-based approach is that, without the requirement of either DNA hydrolysis or shearing, the artificial DNA damage is significantly reduced. Meanwhile, the genomic context of DNA damage can be effectively detected.
The only major technical challenge to single-cell WGA is the large number of amplification errors, which could limit the accuracy in single-cell WGA based approaches (Zong et al., 2012; Chen et al., 2017; Luquette et al., 2019; Bohrson et al., 2019; Xing et al., 2021). In LCS-WGA, we have overcome this technical hurdle by employing linear copying and then a split-amplification scheme. Briefly, we first perform three cycles of low-temperature annealing and extension with temperature ramping to generate multiple copies of the genomic DNA of the single cell as the preamplification step (Figure 1A). In this process, the DNA copies directly generated from the original genomic DNA are termed as semiamplicons, and the DNA products that are copied from the semiamplicons are termed full amplicons. It is worth pointing out that the semiamplicons are linearly produced original genomic DNA of the single cell. The full amplicons are nonlinearly copied from semiamplicons. To quench the nonlinearly produced full amplicons from amplification in the downstream multiple displacement amplification (MDA) reaction, we perform a double-stranded conversion, to convert all the full amplicons into double-stranded DNA, while the semiamplicons are kept as single-stranded DNA. Next, we split the preamplification products into three tubes, to perform independent MDA reactions. As MDA only amplifies single-stranded DNA, the double-stranded full amplicons cannot be amplified and, as a result, only linearly copied DNA products and original genomic DNA are amplified in these MDA reactions. Importantly, since the amplification errors in semiamplicons are independently produced during the preamplification step, we can cross-compare the sequencing result of different splits, to effectively filter out amplification errors. Here, we employ a two-split detection criterion, by which we only keep the variants detected in at least two split samples in the variant calling step (Figure 1B). Based on amplification errors in single split reaction, we estimate that the false positive rate due to amplification errors is 8.3 × 10-10 per base when we apply two-split detection criterion (Zhu et al., 2021).
With this new method, we have successfully examined the DNA damage in human neurons and identified the association between the non-uniform distribution of DNA damage and 3D chromatin structure, as well as the connection between high-damage genes and the altered gene expressions in Alzheimer's disease and autism. This method can be readily applied to different biological contexts, to quantify damage levels in cells and, more importantly, identify high-damage genes. We believe that the characterization of the damagenome in different cell types from different tissues could shed new light on complex human diseases.
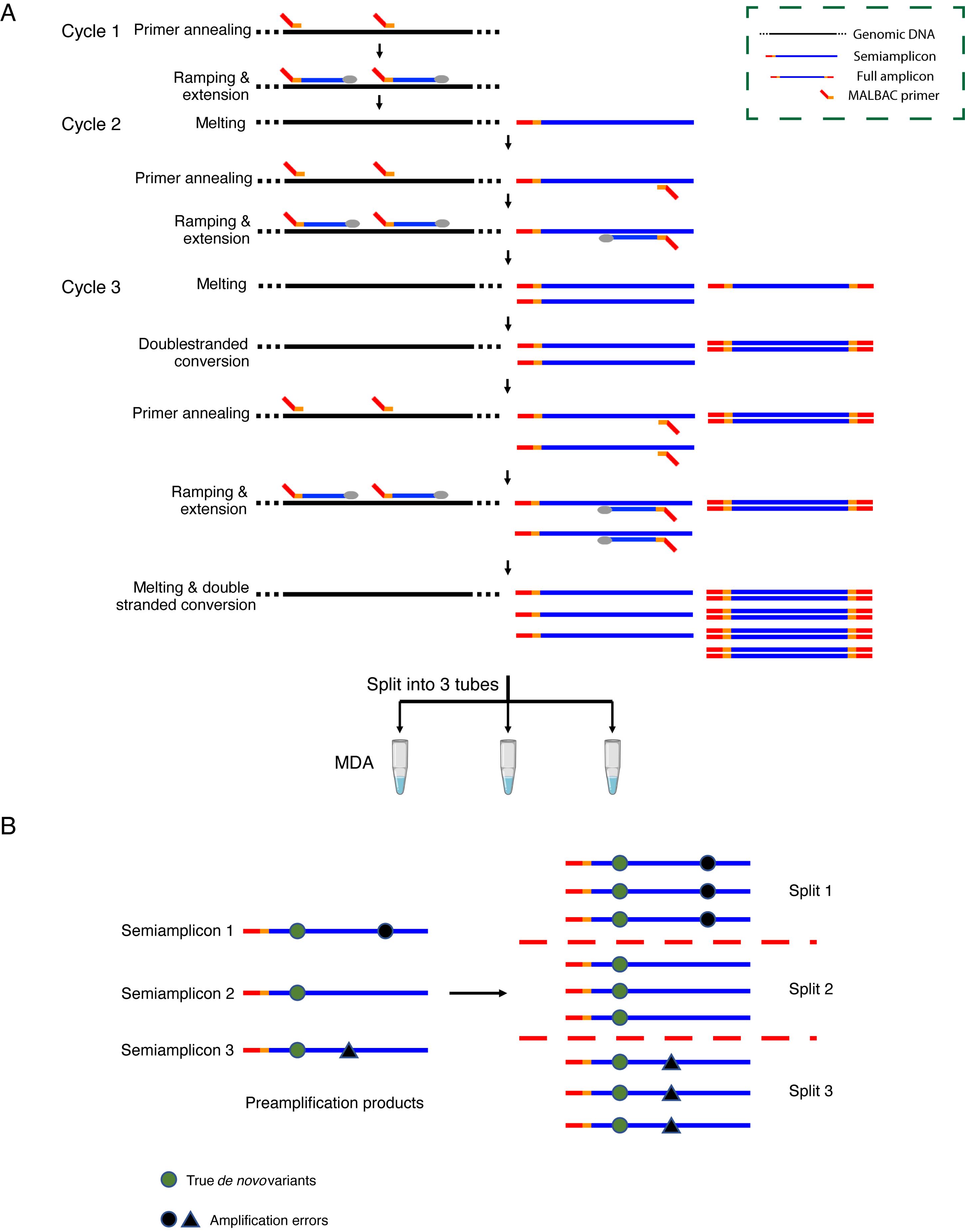
Figure 1. Scheme of LCS-WGA. (A) The scheme of pre-amplification and split-MDA. (B) The scheme to distinguish true de novo variants from amplification errors.
Materials and Reagents
VWR SignatureTM micro aerosol filter pipette tips, Volume: 0.1–10 μL (VWR, catalog number: 53509-138)
0.2 mL PCR tubes with Flat Cap (Axygen, catalog number: PCR-02-L-C)
1.7 mL microtube (Olympus plastics, catalog number: 24-282LR)
Notes:
Pipette tips used in this protocol should be low-retention, RNase-free, DNase-free, with aerosol filter.
PCR tubes and microtubes used in this protocol should be low-retention, RNase-free, and DNase-free.
Potassium hydroxide solution, 8 N (Sigma-Aldrich, catalog number: P4494)
DTT, 5 g (Sigma-Aldrich, catalog number: GE17-1318-02)
Ethylenediaminetetraacetic acid (EDTA) solution, 0.5 M in H2O (Sigma-Aldrich, catalog number: E7889)
Tris-HCl, pH = 7.5, 1 M (Invitrogen, catalog number: 15567-027)
Hydrochloric Acid Solution, 2 N (Fisher Scientific, catalog number: SA431)
InvitrogenTM UltraPureTM DEPC-Treated Water (Invitrogen, catalog number: 750024)
ThermoPol Reaction Buffer, 10× (New England BioLabs, catalog number: B9004S)
Uracil-DNA Glycosylase, 5,000 units/mL (New England BioLabs, catalog number: M0280S)
Deoxynucleotide (dNTP) Solution Mix (10 mM) (New England BioLabs, catalog number: N0447S)
Bst DNA Polymerase, Large Fragment (New England BioLabs, catalog number: M0275S)
iTaq Universal SYBR Green Supermix (Bio-Rad, catalog number: 1725124)
phi29 DNA Polymerase, supplemented with phi29 DNA polymerase reaction buffer (10×) and BSA, Molecular Biology Grade (New England BioLabs, catalog number: M0269S)
TWEEN® 20, for molecular biology, viscous liquid (Sigma-Aldrich, catalog number: P9416)
Agencourt AMPure XP (Beckman Coulter, catalog number: A63880)
Illumina Tagment DNA TDE1 Enzyme and Buffer Kit (Illumina, catalog number: 20034197)
Magnesium acetate (Mg(Ac)2) solution, BioUltra, for molecular biology (Sigma-Aldrich, catalog number: 63052)
NEBNext® UltraTM II Q5® Master Mix (New England BioLabs, catalog number: M0544S)
Agilent D1000 ScreenTape (Agilent, catalog number: 5067- 5582)
Agilent D1000 Reagents (Agilent, catalog number: 5067- 5583)
QubitTM dsDNA HS Assay Kit (Invitrogen, catalog number: Q32851)
Phosphate-Buffered Saline (PBS), 1× without calcium and magnesium (Corning, catalog number: 21-040-CV)
DNA oligos (Ordered from Integrated DNA Technologies)
MCF 10A cell line (ATCC CRL-10317)
Dulbecco's Modified Eagle Medium/Nutrient Mixture F-12 (DMEM/F-12), HEPES (Gibco, catalog number: 11330032)
Horse serum (Gibco, catalog number: 16050122)
Hydrocortisone (Sigma-Aldrich, catalog number: H0888)
Cholera toxin (Sigma-Aldrich, catalog number: C8052)
Insulin (Sigma-Aldrich, catalog number: I1882)
Recombinant human epidermal growth factor (ThermoFisher Scientific, catalog number: PHG0311)
Frozen human brain tissue (Obtained from NIH NeuroBioBank: https://neurobiobank.nih.gov/)
Lysis buffer (400 mM KOH, 100 mM DTT, 2 mM EDTA) (see Recipes)
Stop solution (400 mM HCl, 600 mM Tris-HCl) (see Recipes)
UDG reaction mix (see Recipes)
Pre-amplification primer mix (see Recipes)
MDA reaction mix (see Recipes)
Library amplification mix (see Recipes)
UV-treated UltraPure H2O (see Recipes)
Growth medium for the MCF 10A cell line (see Recipes)
Equipment
S1000TM Thermal Cycler (Bio-Rad)
CFX96 Touch Real-Time PCR Detection System (Bio-Rad)
Qubit® 2.0 fluorometer (ThermoFisher)
4200 TapeStation System (Agilent)
CL-1000 Ultraviolet Crosslinker
Microscope Slides (VWR VistaVisionTM HistoBond®, catalog number: 16004-406)
Motorized inverted research microscope (Olympus IX81)
VWR® Mini Centrifuge
GILSON® Pipetman L P2L, 0.2–2 μL, Metal Ejector (for small volume pipetting)
Bunsen burner (natural gas)
Borosilicate glass with filament (Sutter Instrument, catalog number: BF100-58-15)
Aspirator tube assemblies for calibrated microcapillary pipettes (Sigma-Aldrich, catalog number: A5177-5EA)
High-performance computing cluster (Memory: 256 GB, Nodes: 32, Iris 2281T nodes from or similar nodes from Penguin Computing)
Software
Burrows-Wheeler Aligner (BWA, version: 0.7.12-r1039, http://bio-bwa.sourceforge.net/, free)
Genome Analysis Toolkit (GATK, version: 3.8, https://gatk.broadinstitute.org/hc/en-us, free)
Samtools (v1.7, http://www.htslib.org/, free)
Bedtools (v2.17.0, https://bedtools.readthedocs.io/en/latest/, free)
Procedure
文章信息
版权信息
© 2022 The Authors; exclusive licensee Bio-protocol LLC.
如何引用
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Niu, Y., Zhu, Q. and Zong, C. (2022). Single-cell Damagenome Profiling by Linear Copying and Splitting based Whole Genome Amplification (LCS-WGA). Bio-protocol 12(6): e4357. DOI: 10.21769/BioProtoc.4357.
- Zhu, Q., Niu, Y., Gundry, M. and Zong, C. (2021). Single-cell damagenome profiling unveils vulnerable genes and functional pathways in human genome toward DNA damage. Sci Adv 7(27). DOI: 10.1126/sciadv.abf3329.
分类
癌症生物学 > 基因组不稳定性及突变
细胞生物学 > 单细胞分析 > 流式细胞术
分子生物学 > DNA > DNA 测序
您对这篇实验方法有问题吗?
在此处发布您的问题,我们将邀请本文作者来回答。同时,我们会将您的问题发布到Bio-protocol Exchange,以便寻求社区成员的帮助。