

Modeling Perturbations in Protein Filaments at the Micro and Meso Scale Using NAMD and PTools/Heligeom
用NAMD和PTools/Heligeom模拟蛋白质丝的微尺度和中尺度扰动
发布: 2021年07月20日第11卷第14期 DOI: 10.21769/BioProtoc.4097 浏览次数: 3623
评审: Mostafa RahnamaRAMESH KUDIRASonal Patel PatelAnonymous reviewer(s)
参见作者原研究论文
The authors used this protocol in:

Sep 2019
Advertisement










Abstract
Protein filaments are dynamic entities that respond to external stimuli by slightly or substantially modifying the internal binding geometries between successive protomers. This results in overall changes in the filament architecture, which are difficult to model due to the helical character of the system. Here, we describe how distortions in RecA nucleofilaments and their consequences on the filament-DNA and bound DNA-DNA interactions at different stages of the homologous recombination process can be modeled using the PTools/Heligeom software and subsequent molecular dynamics simulation with NAMD. Modeling methods dealing with helical macromolecular objects typically rely on symmetric assemblies and take advantage of known symmetry descriptors. Other methods dealing with single objects, such as MMTK or VMD, do not integrate the specificities of regular assemblies. By basing the model building on binding geometries at the protomer-protomer level, PTools/Heligeom frees the building process from a priori knowledge of the system topology and enables irregular architectures and symmetry disruption to be accounted for.
Graphical abstract:
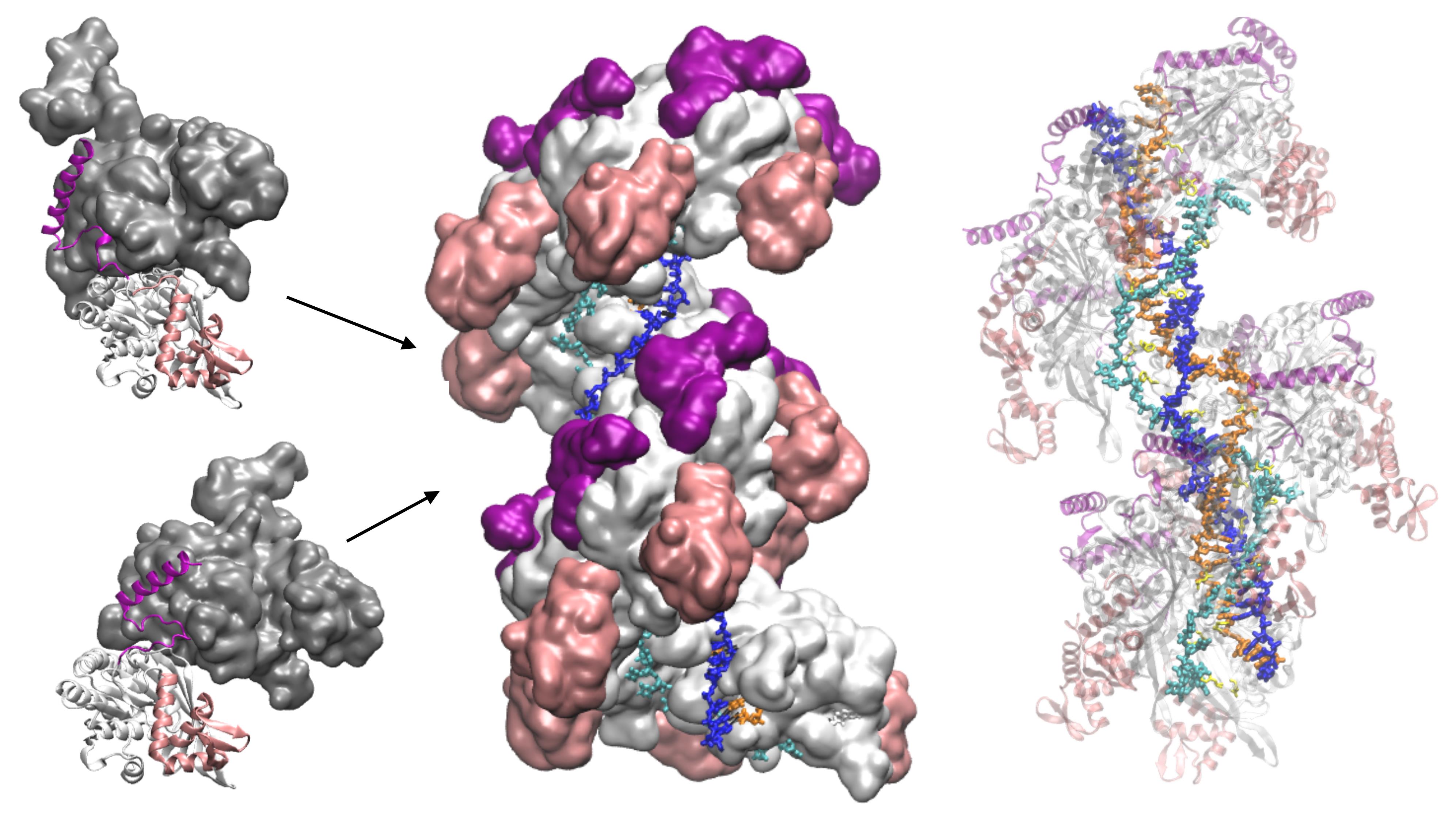
Model of ATP hydrolysis-induced distortions in the recombinant nucleoprotein, obtained by combining RecA-DNA and two RecA-RecA binding geometries.
Background
Oligomeric filamentous assemblies, made of the quasi-regular repetition of macromolecular protomers, are essential components of the cell. Filaments from the cytoskeleton support cell walls or form intracellular transport networks. Other types of filaments are involved in the maintenance, segregation, and repair of DNA (Ghosal and Löwe, 2015). These processes involve dynamic interactions between two generalized helices, the DNA helix and the filament helix, generally accompanied by changes in the DNA linking number and sometimes with coordinated changes in the filament topology. Elucidation of the structure of protein filaments using structural biology methods such as X-ray crystallography or Nuclear Magnetic Resonance has long been highly challenging due to their large size and difficulty in forming crystals. With the recent advances in Cryo-Electron Microscopy, many filament structures are being unveiled; however, filament plasticity and dynamic events are still difficult to capture, and model building remains necessary to decipher how filaments respond to external stimuli. Methods that have been proposed for the modeling of filaments are mostly based on specific docking approaches (Inbar et al., 2005; Casciari et al., 2006; Karaca et al., 2010; Esquivel-Rodriguez et al., 2012) and generally take advantage of known symmetry characteristics (Eisenstein et al., 1997; Berchanski et al., 2003 and 2005; Comeau and Camacho, 2005; Pierce et al., 2005; Schneidman-Duhovny et al., 2005). These methods are not appropriate for investigating structural perturbations in the filament, either global (changes in helical characteristics) or local (symmetry disruption), that appear as responses to external stimuli.
The Heligeom module (Boyer et al., 2015) of the PTools library software (Saladin et al., 2009) has been developed to meet the challenge of modeling helical protein filaments, their interactions with other molecules such as DNA, and their structural responses to external stimuli, either diffuse (changes in longitudinal or torsional stress, following changes in salt concentration or pH), or focused (local molecule binding or interface modifications). Heligeom derives the geometry of oligomeric assemblies from the binding geometry between pairs of neighboring protomers, which offers the possibility to combine several binding modes within the same filament. The PTools library itself gathers a collection of tools to manipulate macromolecules or selected regions of these macromolecules in atomic or coarse-grained representation. While it shares several functionalities with other libraries such as MMTK (Hinsen, 2000) or the VMD Script Library (Humphrey et al., 1996), certain functionalities such as the easy use of screw transformations make it particularly well suited to dealing with helical or quasi-helical assemblies.
Here, we present the application of PTools/Heligeom to model an irregular form of the RecA nucleofilament complex that is active in homologous recombination and includes one to three DNA strands (Boyer et al., 2015). Distortion in the filament results from the introduction of an alternative binding geometry of RecA-RecA interaction at the center of a two-turn helical nucleofilament (twelve monomers). Whereas the principal RecA-RecA binding mode corresponds to the favorable binding geometry between two RecA monomers in the presence of ATP, the alternative binding mode corresponds to the geometry in the presence of ADP. Both binding geometries have been solved by crystallography. The model therefore represents the result of one ATP molecule being hydrolyzed in the filament center. All three DNA strands participate in a strand exchange process. Strand 1 is a damaged DNA strand on which the filament has polymerized; it is bound to DNA-binding site I in the filament. Strand 3 is homologous to strand 1 and occupies the secondary binding site (site II). Strand 2 is complementary to both strand 1 and strand 3. During the strand exchange process catalyzed by the filament, strand 1 captures strand 2, which was initially paired with strand 1. We model three states of the system: the initial state, where only strand 1 is bound to site I (one bound strand); the annealed state, where strand 2 has paired with strand 1 at site I (two bound strands); and the state resulting from strand exchange, where strands 1 and 2 are at site I and strand 3 is at site II (three bound strands). The protocol presented here can be used for any oligomeric system in which three-dimensional structures are available for at least two protein-protein binding geometries obtained under two different conditions, for example, with different cofactors, and present regions of structural similarity. The protocol enables exploration of transient intermediate geometries that are difficult to access experimentally. It is particularly well suited to the study of "collaborative protein filaments," as described in Ghosal and Lowe (2015).
Equipment
Linux workstation (Dell) running a Linux distribution Ubuntu 18.04.5 LTS (GNU/Linux 4.15.0-118-generic x86_64)
Access to a scalar supercomputer (petaflop performance) for molecular dynamics simulations
Software
PTools (Saladin et al., 2009) is a python/C++ library dedicated to the manipulation, assembly, and analysis of macromolecular complexes. It is freely available in the GitHub repository at the address https://github.com/ptools/ptools/tree/develop; it is licensed under the GNU General Public License v3.0. The Heligeom module relates protomer-protomer binding geometries to the geometry of large oligomeric assemblies (Boyer et al., 2015). Heligeom basic functions consist of analyzing protomer interfaces in terms of the helical parameters (Figure 1A) of associated screw objects – pitch (P), number of monomers per turn (N), handedness, see Figure 1A, right – and constructing helices with the desired number of protomers. The helix can optionally be aligned to the Z axis at the origin. Figure 1B shows an example of the workflow of PTools/Heligeom functions that are commonly used in this work. These include selection functions (chains, residue numbers or types, atoms numbers or types), the superposition function that outputs a transformation matrix, and the mat_trans_to_screw function that produces the screw object corresponding to the transformation matrix.
The NAMD software (Scalable Molecular Dynamics, Mackerell et al., 2004; Phillips et al., 2005; http://www.ks.uiuc.edu/Research/namd/) is used to perform energy minimization and molecular dynamics (MD) simulations.
VMD (Humphrey et al., 1996) is a molecular visualization program: VMD can display and analyze large biomolecular systems and their MD trajectories using 3-D graphics and built-in scripting https://www.ks.uiuc.edu/Research/vmd/script_library/.
NAMD and VMD are both developed at the NIH Center for Macromolecular Modeling & Bioinformatics, University of Illinois at Urbana-Champaign, by the Theoretical and Computational Biophysics Group. Both are distributed free of charge with source code.
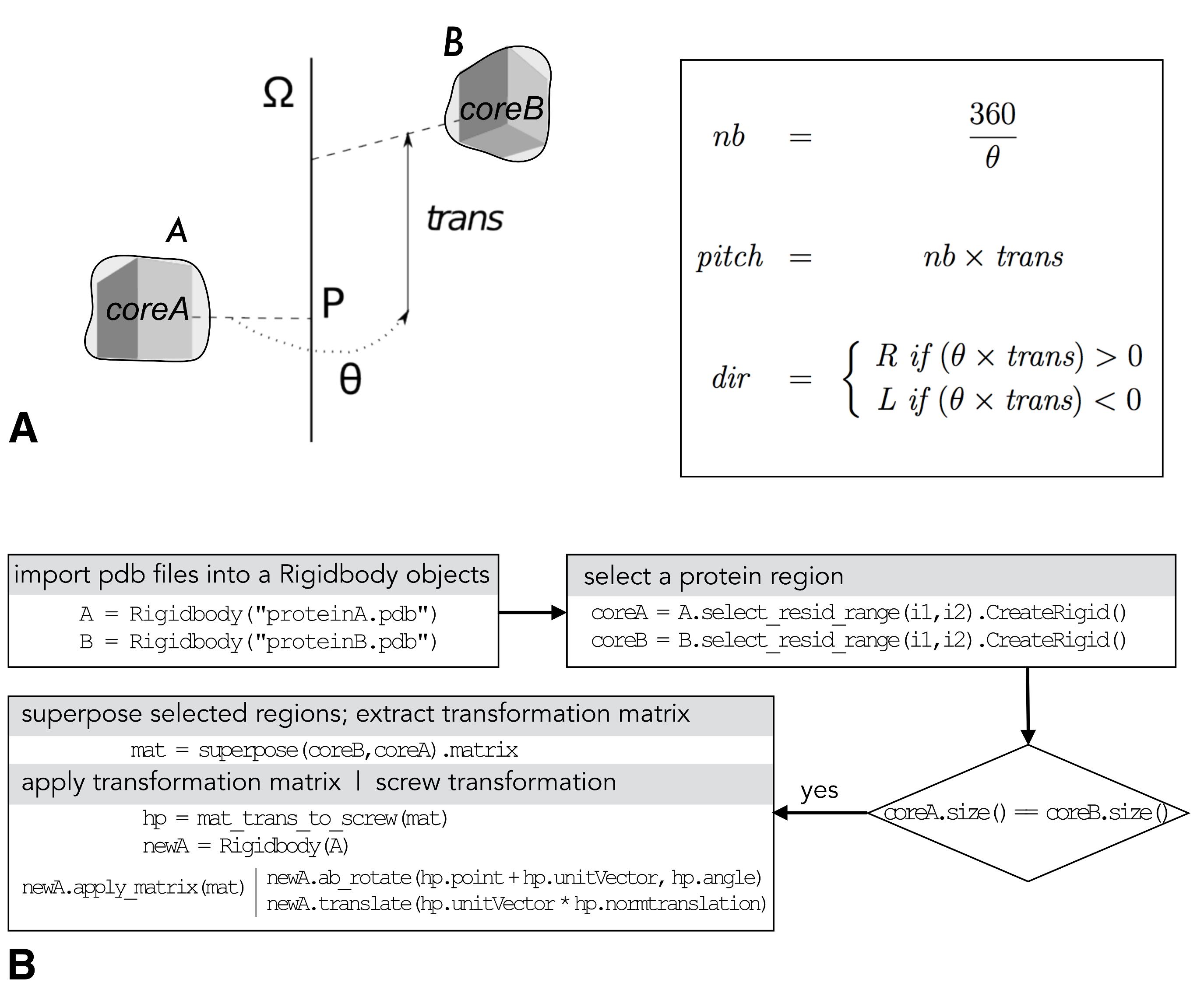
Figure 1. PTools/Heligeom. (A) Left: scheme of a screw transformation between two proteins A and B, with structurally similar core regions coreA and core2; right: calculation of the number of monomers per turn (nb), the pitch and the handedness (R: right, L: left) from the screw parameters θ and trans. (B) Typical PTools operations that enable the combination of available binding geometries. This includes creating Rigidbody objects from the .pdb files ("import .pdb files…"), defining structurally superposable regions with identical sizes using the select_resid_range attribute ("select a protein region"), defining mat, the transformation matrix that superposes the defined regions using the superpose function ("superpose selected regions…"), and defining the screw transformation hp equivalent to mat; example of how to use the screw object hp to create a new Rigidbody object according to the desired screw transformation is given under "apply transformation matrix…"
Procedure
文章信息
版权信息
© 2021 The Authors; exclusive licensee Bio-protocol LLC.
如何引用
Boyer, B., Laurent, B., Robert, C. H. and Prévost, C. (2021). Modeling Perturbations in Protein Filaments at the Micro and Meso Scale Using NAMD and PTools/Heligeom. Bio-protocol 11(14): e4097. DOI: 10.21769/BioProtoc.4097.
分类
生物物理学 > 大分子模拟
生物化学 > 蛋白质 > 结构
分子生物学 > DNA
您对这篇实验方法有问题吗?
在此处发布您的问题,我们将邀请本文作者来回答。同时,我们会将您的问题发布到Bio-protocol Exchange,以便寻求社区成员的帮助。