

Evaluation of the Sequence Variability within the PCR Primer/Probe Target Regions of the SARS-CoV-2 Genome
SARS-CoV-2基因组PCR引物/探针靶区序列变异性评价
发布: 2020年12月20日第10卷第24期 DOI: 10.21769/BioProtoc.3871 浏览次数: 5958
评审: Vasudevan AchuthanRan ChenChhuttan L MeenaAnonymous reviewer(s)
参见作者原研究论文
The authors used this protocol in:

Jun 2020
Advertisement










Abstract
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2; initially named 2019-nCoV) is responsible for the recent coronavirus disease (COVID-19) pandemic, and polymerase chain reaction (PCR) is the current standard method for diagnosis from patient samples. As PCR assays are prone to sequence mismatches due to mutations in the viral genome, it is important to verify the genomic variability at primer/probe binding regions periodically. This step-by-step protocol describes a bioinformatics approach for an extensive evaluation of the sequence variability within the primer/probe target regions of the SARS-CoV-2 genome. The protocol can be applied to any molecular diagnostic assay of choice using freely available software programs and the ready-to-use multiple sequence alignment (MSA) file provided.
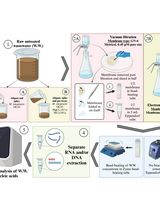
Graphic abstract: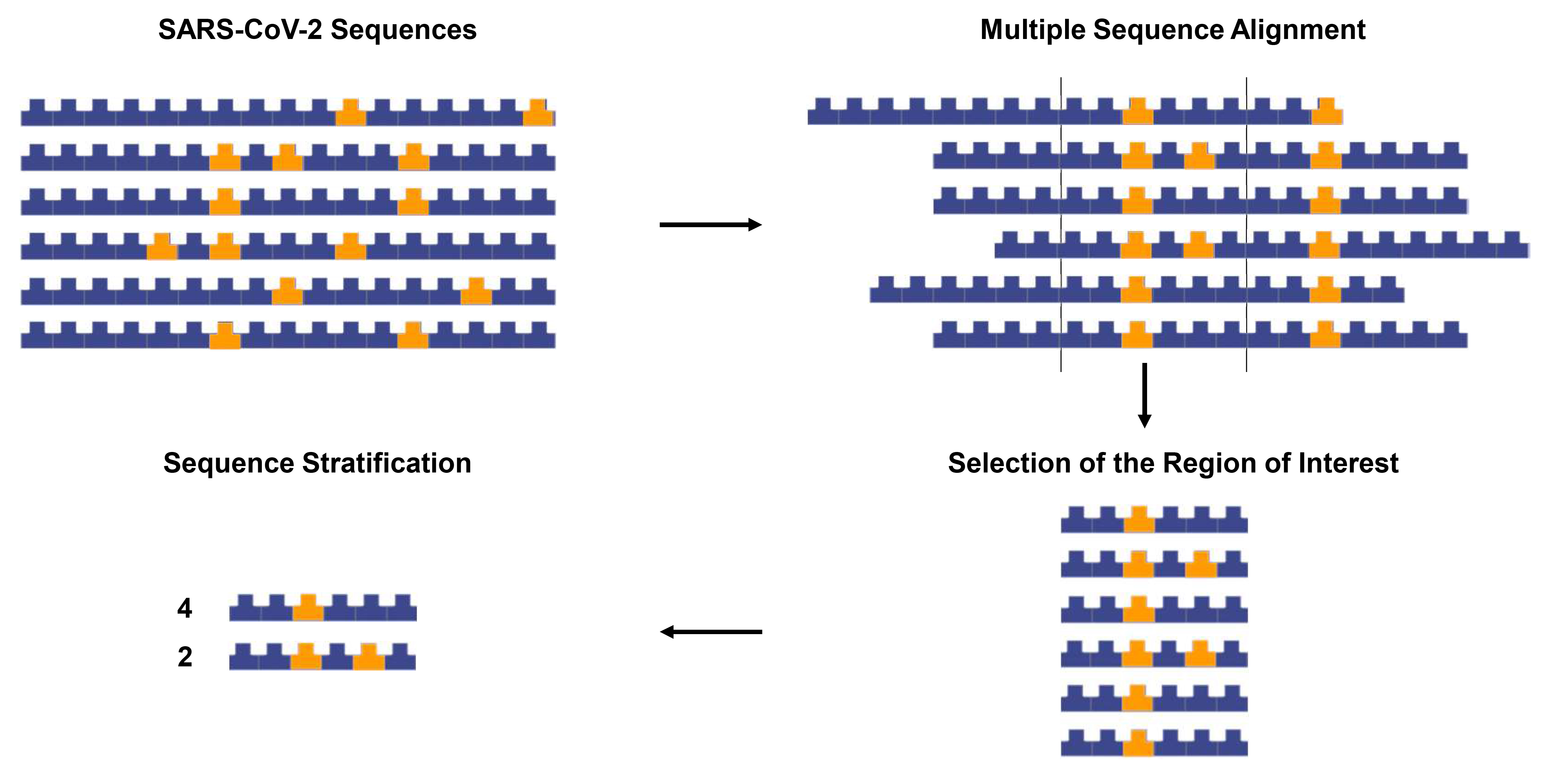
Overview of the sequence tracing protocol. The figure was created using the Library of Science and Medical Illustrations from somersault18:24 licensed under a CC BY-NC-SA 4.0 license (https://creativecommons.org/licenses/by-nc-sa/4.0/).
Video abstract: https://youtu.be/M1lV1liWE9k
Background
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2; initially named as 2019-nCoV) is the cause of novel coronavirus disease termed COVID-19. The virus originated from Wuhan, China and rapidly spreaded around the world causing a global pandemic (Worldometers.info, 2020). Sequencing of the virus showed that its single-stranded RNA genome is ~30 kb in size (Chan et al., 2020; Lu et al., 2020; Wu et al., 2020; Zhou et al., 2020). Availability of the viral sequence early in the outbreak helped the development of several polymerase chain reaction (PCR) detection protocols that have been instrumental in the diagnosis of the disease from patients samples (WHO, 2020). However, genetic variability in the viral genome during natural evolution poses a potential risk of mismatches between the diagnostic assays and the template that can result in false-negative results (Whiley and Sloots, 2005; Chow et al., 2011). Sequences of SARS-CoV-2 viruses isolated from around the world are being deposited in the sequence databases and mutations have been identified in the genomes of the circulating viruses (Ugurel et al., 2020).
We performed an extensive evaluation of published diagnostic PCR assays, including those recommended by the World Health Organization (WHO), based on evaluation of sequence variation in the primer/probe binding regions using more than 17,000 publicly available viral sequences (Khan and Cheung, 2020). Another concurrent publication reported mutations in primer/probe binding regions using 1825 sequences but a detailed sequence tracing protocol was not provided (Osorio and Correia-Neves, 2020). This step-by-step protocol outlines a bioinformatics pipeline that uses freely available open-source software programs. The pipeline can be performed on a regular desktop computer without any need for special hardware and does not require extensive computational skills. The provision of a ready-to-use Multiple Sequence Alignment (MSA) file through Open Science Framework (OSF) makes it an even more intuitive task. Inclusivity analysis through verification of in silico nucleotide identity match is one of the regulatory requirements for approval of COVID-19 diagnostic assays (Commission-Services, 2020; FDA, 2020; Health-Canada, 2020). The protocol can also be applied to other molecular diagnostic assays of SARS-CoV-2 including point-of-care CRISPR-based diagnostic assays under development (Tsang and LaManna, 2020).
Equipment
A regular Windows or Mac OS X laptop or desktop.
Note: There is no specific processor or RAM requirement, but memory issues can be avoided by opening a limited number of files at the same time. The outlined protocol was performed on a laptop installed with Windows 10, an Intel Core i5-8265U processor, CPU @1.60GHz and an 8 GB RAM.
Software
MAFFT version 7 online service (Katoh et al., 2002 and 2019) (available from https://mafft.cbrc.jp/alignment/software/closelyrelatedviralgenomes.html)
AliView version 1.26 (Larsson, 2014) (available from https://ormbunkar.se/aliview/)
Sequence Manipulation Suite version 2 (Stothard, 2000) (available from https://www.bioinformatics.org/sms2/rev_comp.html)
SequenceTracer (Nagy et al., 2019) (available from http://www1.szu.cz:8080/EntropyCalcWeb/sequences)
ElimDupes (https://www.hiv.lanl.gov/content/sequence/elimdupesv2/elimdupes.html)
PNNS calculator (available from http://entropy.szu.cz:8080/EntropyCalcWeb/pnns)
A web browser (for example Google Chrome or Mozilla Firefox)
A text editor (for example Microsoft Notepad)
Procedure
文章信息
版权信息
© 2020 The Authors; exclusive licensee Bio-protocol LLC.
如何引用
Khan, K. A. and Cheung, P. (2020). Evaluation of the Sequence Variability within the PCR Primer/Probe Target Regions of the SARS-CoV-2 Genome. Bio-protocol 10(24): e3871. DOI: 10.21769/BioProtoc.3871.
分类
微生物学 > 病原体检测 > PCR
分子生物学 > DNA > PCR
您对这篇实验方法有问题吗?
在此处发布您的问题,我们将邀请本文作者来回答。同时,我们会将您的问题发布到Bio-protocol Exchange,以便寻求社区成员的帮助。