

Whole-genome Identification of Transcriptional Start Sites by Differential RNA-seq in Bacteria
细菌转录起始位点的差异RNA序列全基因组鉴定

发布: 2020年09月20日第10卷第18期 DOI: 10.21769/BioProtoc.3757 浏览次数: 7208
评审: Imre GáspárShyam SolankiAdam Idoine
参见作者原研究论文
The authors used this protocol in:

Apr 2020
Advertisement









Abstract
Gene transcription in bacteria often starts some nucleotides upstream of the start codon. Identifying the specific Transcriptional Start Site (TSS) is essential for genetic manipulation, as in many cases upstream of the start codon there are sequence elements that are involved in gene expression regulation. Taken into account the classical gene structure, we are able to identify two kinds of transcriptional start site: primary and secondary. A primary transcriptional start site is located some nucleotides upstream of the translational start site, while a secondary transcriptional start site is located within the gene encoding sequence.
Here, we present a step by step protocol for genome-wide transcriptional start sites determination by differential RNA-sequencing (dRNA-seq) using the enteric pathogen Shigella flexneri serotype 5a strain M90T as model. However, this method can be employed in any other bacterial species of choice. In the first steps, total RNA is purified from bacterial cultures using the hot phenol method. Ribosomal RNA (rRNA) is specifically depleted via hybridization probes using a commercial kit. A 5′-monophosphate-dependent exonuclease (TEX)-treated RNA library enriched in primary transcripts is then prepared for comparison with a library that has not undergone TEX-treatment, followed by ligation of an RNA linker adaptor of known sequence allowing the determination of TSS with single nucleotide precision. Finally, the RNA is processed for Illumina sequencing library preparation and sequenced as purchased service. TSS are identified by in-house bioinformatic analysis.
Our protocol is cost-effective as it minimizes the use of commercial kits and employs freely available software.
Background
Transcription in bacteria is initiated by the RNA polymerase holoenzyme, which recognizes specific sequence elements on the DNA within the promotor region, to which sigma factors are bound (Feklistov et al., 2014). This RNA polymerase holoenzyme binding site defines the Transcriptional Start Site and the direction of transcription. For example, the most common house-keeping sigma factor, named 𝝈70 in Escherichia coli, recognizes two elements centered approximately 10 and 35 bp upstream of the TSS (Feklistov et al., 2014). The RNA polymerase holoenzyme melts the double stranded DNA between 11 nt upstream (position -11) to 3 nt downstream (+3) of the TSS (+1), and the single-stranded DNA can then be used as template for the addition of tri-phosphorylated ribonucleotides. The initiation starts mainly at a specific position, but sometimes “wobbles” of one or more bases up- or downstream are encountered (Murakami and Darst, 2003; Robb et al., 2013; Vvedenskaya et al., 2015). The DNA sequence around TSS have long been recognized as crucial for gene regulation in bacteria (Jacob and Monod, 1961). Depending on the position within the gene structure, which begins with a start codon (usually ATG) and finishes with one of the three stop codons, we can identify two types of transcriptional start sites: primary and secondary. Primary transcriptional start sites (pTSS) are located some nucleotides upstream of the translational start site, while the secondary transcriptional start sites (sTSS) are located within the gene encoding sequence (Figure 1).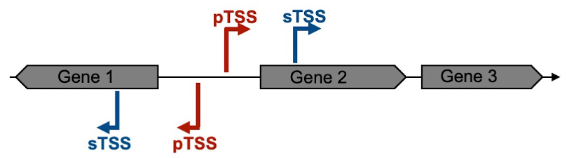
Figure 1. Schematic representation of the Primary and Secondary Transcriptional Start Site definition
Until the advent of next-generation sequencing, in order to locate the TSS of a specific RNA, it was necessary to examine each transcript individually, using either the S1 protection assay, primer extension or a 5’ RACE method (Sharma and Vogel, 2014). Owing to the increasing popularity and a decrease in costs of high-through put sequencing, in 2010 differential RNA-seq (dRNA-seq) was developed to simultaneously map all TSS of a genome using Helicobacter pylori as first model organism (Sharma et al., 2010). Since then, this method has been widely employed to determine the TSS of several bacterial species (Berghoff et al., 2009; Jager et al., 2009; Albrecht et al., 2010 and 2011; Bohn et al., 2010; Irnov et al., 2010; Schluter et al., 2010; Sharma et al., 2010; Beckmann et al., 2011; Deltcheva et al., 2011; Filiatrault et al., 2011; Mitschke et al., 2011a and 2011b; Kroger et al., 2012 and 2013; Madhugiri et al., 2012; Ramachandran et al., 2012 and 2014; Sahr et al., 2012; Schmidtke et al., 2012; Wilms et al., 2012; Cortes et al., 2013; Dugar et al., 2013; Mentz et al., 2013; Nickel et al., 2013; Pfeifer-Sancar et al., 2013; Porcelli et al., 2013; Schluter et al., 2013; Voss et al., 2013; Wiegand et al., 2013; Zhang et al., 2013; Voigt et al., 2014; Cervantes-Rivera et al., 2020).
Primary transcripts of prokaryotes carry a triphosphate at their 5’-ends. In contrast, processed or degraded RNAs only carry a monophosphate at their 5’-ends. This is also the case of ribosomal RNA (rRNA) (Schoenberg, 2007). The dRNA-seq approach used here exploits the properties of a 5’-monophosphate-dependent exonuclease (TEX) to selectively degrade processed transcripts, thereby enriching for unprocessed RNA species carrying a native 5’-triphosphate (Schoenberg, 2007). TSS can then be identified by comparing TEX-treated and untreated RNA-seq libraries, where TSS appear as localized maxima in coverage enriched upon TEX-treatment (Sharma et al., 2010).
Until 2013 TSS annotation was performed manually, but this method is arduous and time-consuming. Nowadays many computational tools are available for automatic TSS annotation using dRNA-seq data. These include TSSPredator (Dugar et al., 2013), TSSAR (Amman et al., 2014), TruHMM (Li et al., 2013), TSSer (Jorjani and Zavolan, 2014) and ReadXplorer2 (Hilker et al., 2016).
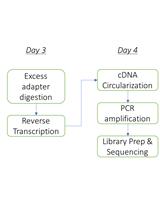
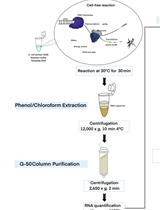
Here, we present a step by step protocol for TSS determination through comparison of TEX-treated and untreated RNA libraries in Shigella flexneri serotype 5a strain M90T as originally performed in (Cervantes-Rivera et al., 2020). The overall workflow is illustrated in Figure 2.
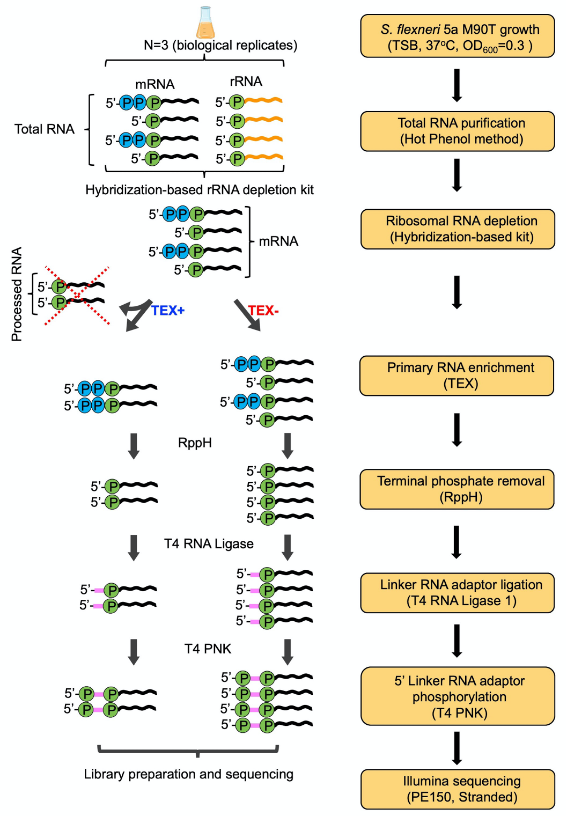
Figure 2. Workflow of dRNA-seq for whole-genome Transcriptional Start Sites identification
Materials and Reagents
- Sterile syringe filter with pore size 0.22 μm (Merck, catalog number: SLGV033RK )
- Nitrile gloves
- Culture tube of 13 ml (TPP, catalog number: 91016 )
- Microfuge tubes 1.5 ml (Eppendorf, Safe-Lock tubes, catalog number: 00 30120086 )
Note: In principle all prepackaged tubes on the market are RNase free and can be used. - Centrifuge tubes of 50 ml (Sarstedt, catalog number: 62.547.254 )
- Pipette tips (VWR, catalog numbers: 89041-404 , 89041-412 , 89041-400 )
Note: In principle all prepackaged tips on the market are RNase free and can be used. - Semi-micro cuvette (Sarstedt, catalog number: 67.742 )
- Shigella flexneri 5a M90T (Sansonetti et al., 1982), can also be purchased from ATCC and available on request from the authors
- Tryptone soy broth (TSB) ready to use powder (Merck, catalog number: 105459 )
- Tryptone soy agar (TSA) ready to use powder (Merck, catalog number: 105458 )
- Congo red (Sigma, catalog number: C6277 )
- Diethyl pyrocarbonate (DEPC) (Sigma, catalog number: 40718 )
- Sodium dodecyl sulfate (SDS) (Sigma, catalog number: L3771 )
- Bromophenol blue sodium salt (Sigma, catalog number: B8026 )
- Xylene cyanol FF (Sigma, catalog number: X4126 )
- Glycerol (Sigma, catalog number: G5516 )
- Formamide (Sigma, catalog number: 47671 )
- Ethanol absolute (VWR Chemicals, catalog number: 20821.558 )
- Sodium acetate 3 M, pH 5.5 (Ambion, catalog number: AM9740 ).
- EDTA, disodium salt, dihydrate (Sigma, catalog number: 324503 )
- Sodium hydroxide (Sigma, catalog number: S8045 )
- Trizma base (Sigma, catalog number: T1503 )
- Acetic acid (Sigma, catalog number: 695092 )
- Agarose (VWR Life Science, catalog number: 35-1020 )
- DNase I 10 U/ml (Roche, catalog number: 0 4716728001 )
- RNaseOUT 40 U/ml (Invitrogen, catalog number: 10777019 )
- Chloroform (Sigma, Catalog number: C2432 )
- Phenol solution pH 4.3 ± 0.2 (Sigma, catalog number: P4682-400ML )
- Isoamyl alcohol (Sigma, catalog number: W205702 )
- Sodium acetate pH 5.5 (Ambion, catalog number: AM9740 )
- Glycogen 20 mg/ml (Invitrogen, catalog number: R0551 )
- Oligos SF-Hfq-F 5′-ACGATGAAATGGTTTATCGAG-3′ and SF-Hfq-R 5′-ACTGCTTTACCTTCACCTACA-3′ (Sigma, custom order)
- GeneRuler 1 kb DNA ladder (Thermo Scientific, catalog number: SM0211 )
- Linker-RNA-adaptor 5′-GACCUUGGCUGUCACUCA-3′ (Sigma, custom order)
- Ribo-Zero rRNA Removal Kit for bacteria (Illumina, catalog number: MRZB12424 )
Note: This product is not available anymore on the market, but can be replaced with Pan-Prokaryote riboPOOL kit (siTOOLS BIOTECH, Pan-Prokaryote v2), RiboCop rRNA Depletion Kit for Bacteria (Lexogen, catalog number: 125-127 ) or any other rRNA depletion method based on the use of hybridization probes. Replacement products relying on enzymatic-based rRNA depletion such as the Ribo-Zero Plus rRNA Depletion Kit (Illumina, catalog number: 20037125 ) are not suitable. - Terminator-5′-Phosphate-Dependent Exonuclease (TEX) (Lucigene, catalog number: TER51020 )
- 5’-Pyrophosphohydrolase 5,000 units/ml (RppH) (New England BioLabs, catalog number: M0356S )
- T4 RNA ligase 10,000 U/ml (New England BioLabs, catalog number: M0204L )
- T4 polynucleotide kinase (PNK) 10,000 U/ml (New England BioLabs, catalog number: M0201L )
- RNaseZap (Ambion, catalog number: AM9780 )
- Dream Taq DNA polymerase (Thermo Scientific, catalog number: EP0701 )
- dNTPs mix, 2 mM each (Thermo Scientific, catalog number: R0241 )
- GelRed nucleic acid gel stain (Biotium, catalog number: 41003 )
- ATP solution (100 mM) (Thermo Scientific, catalog number: R0441 )
- Tryptone soy broth (TSB) medium (see Recipes)
- Tryptone soy agar (TSA) plates (see Recipes)
- Congo red solution (see Recipes)
- Nuclease-free water (see Recipes)
- Sodium dodecyl sulfate (SDS) 10% solution (see Recipes)
- EDTA 0.5 M, pH 8 (see Recipes)
- 6x DNA Loading buffer (see Recipes)
- 2x RNA Loading buffer (see Recipes)
- Stop Solution (see Recipes)
- Ethanol 80% (see Recipes)
- Lysis solution (see Recipes)
- 10x TAE (see Recipes)
- 1% Agarose (see Recipes)
- Phe-CHISAM solution (see Recipes)
- CHISAM (see Recipes)
Equipment
- Microcentrifuge, refrigerated (VWR, model: Micro Star 17R )
- Spectrophotometer (Amersham, model: Ultrospec 2100 pro )
- Pipettes (Eppendorf Research plus)
- Microwave oven (Whirlpool, model: MD101 )
- Centrifuge (Eppendorf, model: 5810R )
- NanoDrop ND-1000 spectrophotometer (Saveen & Werner AB)
- Orbital Shaker (Edmund Bühler, model: Swip SM25 )
- Block heater (VWR, model: 460-0353 )
- DynaMag-2 Magnet (Thermo Fisher Scientific, catalog number: 12321D )
- T100 Thermo cycler (Bio-Rad, catalog number: 186-1096 )
- Chemical hood
- Horizontal gel electrophoresis system (VWR, model: 700-0015 )
- Electrophoresis power supply (VWR, model: E0202 )
Software
Programs
All programs used in this protocol are freely available
- bcl2fastq (https://emea.support.illumina.com/downloads/bcl2fastq-conversion-software-v2-20.html)
- FastQC/0.11.8 (https://www.bioinformatics.babraham.ac.uk/projects/download.html#fastqc) (Wingett and Andrews, 2018)
- MultiQC/1.8 (https://multiqc.info/) (Ewels et al., 2016)
- trimmomatic/0.36 (http://www.usadellab.org/cms/?page=trimmomatic) (Bolger et al., 2014)
- samtools/1.9 (http://www.htslib.org/doc/samtools.html) (Li et al., 2009)
- bowtie2/2.3.5.1 (http://bowtie-bio.sourceforge.net/bowtie2/index.shtml) (Langmead and Salzberg, 2012)
- IGV/2.3.82 (https://software.broadinstitute.org/software/igv/) (Thorvaldsdottir et al., 2013)
- ReadXplorer/2.2.3 (https://www.uni-giessen.de/fbz/fb08/Inst/bioinformatik/software/ReadXplorer) (Hilker et al., 2014; Hilker et al., 2016)
Databases
- GenBank (https://www.ncbi.nlm.nih.gov/genome/182?genome_assembly_id=493862)
- RefSeq (https://www.ncbi.nlm.nih.gov/nuccore/NZ_CP037923.1, https://www.ncbi.nlm.nih.gov/nuccore/NZ_CP037924.1)
- RegulonDB (http://regulondb.ccg.unam.mx/)
- dRNA-seq raw data are available on the SRA database under the accession numbers: SRR8921222(dRNA-Seq_TEX_Positive), SRR8921223 (dRNA-Seq_TEX_Negative) (https://dataview.ncbi.nlm.nih.gov/)
Procedure
文章信息
版权信息
© 2020 The Authors; exclusive licensee Bio-protocol LLC.
如何引用
Cervantes-Rivera, R. and Puhar, A. (2020). Whole-genome Identification of Transcriptional Start Sites by Differential RNA-seq in Bacteria. Bio-protocol 10(18): e3757. DOI: 10.21769/BioProtoc.3757.
分类
微生物学 > 微生物遗传学 > 基因表达
微生物学 > 微生物遗传学 > RNA > 测序
系统生物学 > 基因组学 > 测序
您对这篇实验方法有问题吗?
在此处发布您的问题,我们将邀请本文作者来回答。同时,我们会将您的问题发布到Bio-protocol Exchange,以便寻求社区成员的帮助。