

A Workflow for Ultra-rapid Analysis of Histone Post-translational Modifications with Direct-injection Mass Spectrometry
使用直接进样质谱对组蛋白翻译后修饰进行超快速分析的工作流程

发布: 2020年09月20日第10卷第18期 DOI: 10.21769/BioProtoc.3756 浏览次数: 7083
评审: Rohit JainZijian ZhangAnonymous reviewer(s)
参见作者原研究论文
The authors used this protocol in:

May 2019
Advertisement










Abstract
Chromatin modifications, like histone post translational modifications (PTMs), are critical for tuning gene expression and many other aspects of cell phenotype. Liquid chromatography coupled to mass spectrometry (LC-MS) has become the most suitable method to analyze histones and histone PTMs in a large-scale manner. Selected histone PTMs have known functions, and their aberrant regulation is linked to a wide variety of diseases, including cancer. However, histone analysis is scarcely used in diagnostics, partially due to the limited throughput and not ideal reproducibility of LC-MS based analysis. We describe a workflow that allows for high-throughput sample preparation is less than a day using 96-well plates. Following preparation, samples are sprayed into MS without LC, using an automated direct injection (DI-MS) method. Each analysis provides accurate quantification for 29 peptide sequences with 45 PTMs (methylations, acetylations and phosphorylations) for a total of 151 histone marks plus 16 unmodified histone peptides for relative quantification of histone variants. This workflow allows for < 1 min MS runs and higher reproducibility and robustness due to the absence of carryover or LC-based batch effects. Finally, we describe an engineered peptide sequence used to accurately monitor the efficiency of sample preparation, which can be detected during the DI-MS run.
Keywords: Histone (组蛋白)Background
Histones are basic proteins with a globular head and a N-terminal tail rich in arginine and lysine residues. A pair each of canonical histone H2A, H2B, H3 and H4 (called core histones) form an octomer around which 147 bp of DNA winds to form a nucleosome. The tails from these core histones are extensively modified by covalent post translational modifications (PTMs) including methylations, phosphorylations, and acyl groups including acetylations, sugar derivatives and by hundreds of other groups mostly at the lysine residues (Figure 1) (Zhao and Garcia, 2015). These histone PTMs modulate chromatin compaction by altering the charge at the DNA-histone interface and by acting as docking sites for chromatin-associated proteins.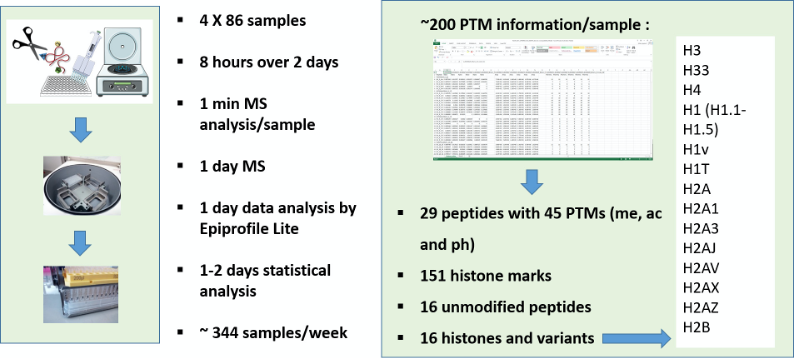
Figure 1. Merits of DI-MS workflow. Multiplexing sample preparation from extraction to derivatization/digestion to desating in a highthroughput manner. Sample preparation still requires at least one day to get histones ready for analysis. However, the workflow allows to multiplex potentially thousands of samples in a single batch, and they can then be analyzed at the rate of one per minute. Virtually, about 344 samples can be analyzed in a regular week (5-days work), when four 96-well plates are processed simultaneously. This capacity is higher than any existing diagnostics facility analyzing histone modifications. Each sample yields data with 220 relative quantifications from 29 peptides, belonging to 16 histone types, both canonical and variant, bearing single or multiple PTMs such as methylations, acetylations and phosphorylations in a total of 45 loci is obtained within a week.
Every eukaryotic cell has a tightly tuned chromatin state, whether it is pluripotent or differentiated, healthy or diseased. Actively transcribed chromatin domains are commonly referred as euchromatin, while silenced sequestered chromatin domains are defined as heterochromatin. While acetylation is generally assigned to accessible chromatin, heterochromatin is associated to more precise histone marks. For instance, methylation of lysine 9 on histone H3 (H3K9me) is the target of the heterochromatin protein 1 (HP1) and it is well known to create constitutive silenced chromatin domains (Nakayama et al., 2001). Other lysine methylations have other biological roles; H3K27 trimethylation is another heterochromatic marker, while methylation of H3K4, H3K36, or H3K79 are associated with active chromatin (Maison and Almouzni, 2004; Peterson and Laniel, 2004; Bannister et al., 2005). Increasingly, we are recognizing cross-talk between different modifications as an extra level of regulation of chromatin function (Kouzarides, 2007). In this context, the ability to differentially quantify e.g., the relative abundance on chromatin of H3K9me3 vs H3K9me3K14ac allows to define the co-existence frequency of histone PTMs.
Conventional antibody-based approaches are very limited for histone PTM analysis due to the following reasons: (i) only very few antibodies recognize multiple PTMs; (ii) only known modifications can be studied with this approach; (iii) antibody-based techniques rarely can be adopted in a large-scale manner, necessary to investigate combinatorial codes; (iv) the specificity of antibodies for histone PTMs, even if commercial, is sometimes not sufficient. Egelhofer and coworkers reported that “more than 25% of commercial antibodies failed specificity tests by dot blot or western blot; among specific antibodies, more than 20% failed in chromatin immunoprecipitation experiments” (Egelhofer et al., 2011). Mass spectrometry (MS) is currently the most suitable analytical tool to study novel and/or combinatorial PTMs, especially due to the high sensitivity, high mass accuracy and the possibility to perform large-scale analyses.
The most widely adopted proteomics workflow for histone analysis is the so-called “bottom-up” approach, where histones are digested into 4-20 amino acid long peptides. We and others have optimized a protocol that allows for the use of trypsin as digestion enzyme (cleaves after K/R) despite the large frequency of arginine (R) and lysine (K) residues on histone proteins, i.e., about 30% of all amino acids. This is then followed up with derivatizing unmodified and monomethylated lysine residues with propionylation (Sidoli et al., 2015) or heavy-labeled acetylation (Bonaldi et al., 2004) to prevent lysine cleavage by trypsin. This yields proper size peptides for best ionization in liquid chromatography coupled online with mass spectrometry (LC-MS). Nevertheless, there are some inherent problems in using LC such as batch effects resulting from column conditions, long gradients to fractionate peptides online, thereby limited sample throughput and unpredictable ionization efficiency for different peptides. While some batch effect arising from performing the sample preparation at different times (even by the same person) and the calibration status of the mass spectrometer still persists, the huge variability that HPLC contributed to MS-aided PTM analysis has been greatly reduced using this DI-MS (Sidoli et al., 2015). Good lab practices such as preparing and analyzing all samples pertaining to a study in a high throughput manner will reduce batch effects. We also recommend ensuring that samples are relatively free of ionizable contaminants as these “background” interferences lead to sample-specific signal suppression in mass spectrometry. Sample clean-up is very crucial as injecting contaminants reduce the radio frequency-guided ion transmission by the S-lens, resulting in poor data acquisition and expensive instrument maintenance costs. In all, DI-MS is straightforward to implement in a mass spectrometry lab or core facility that has the technical expertise for handling LC-MS instruments.
Once high throughput quantitative histone PTM analysis is established as we describe in this protocol, there are exciting opportunities for researchers to examine the missing epigenetic aspects of biological processes and diseases, in the comfort of their own laboratories. Moreover, about 200 pieces of information can be generated for over 300 samples/week making it especially amenable for discovery of epigenetic biomarkers for diseases.
Though DI-MS can be optimized for any analyte including metabolites (Kirwan et al., 2014) and nucleic acids (Quinn et al., 2013), we leveraged a high-throughput infusion workflow for histone PTM analysis because of our familiarity with both the advantages and challenges associated with it as well as the importance for basic science and disease phenotypes. Some properties of histones lend itself for clean extraction, ideal proteolysis and data-independent acquisition (DIA) in the mass spectrometer. The abundance of histones in the nuclear compartment in association with DNA (chromatin) and their basic nature due to arginine and lysine residues have been exploited for extraction, while the specificity of trypsin to the multiple arginine and lysine sites that are unmodified post-translationally provide right-sized (4-20 aminoacids long) peptides for bottom-up strategy. Extracting histones from cell lysates after RIPA or other common cell lysis buffers is not ideal for histones quantification; therefore, we combined cell lysis with nuclear fractionation to extract the chromatin-bound histones. This chromatin-enriched fraction represents 11% of the total cellular proteins and consists predominantly of histone proteins (H1, H2A, H2B, H3, and H4) and other minor components (Shiio et al., 2003). Though chromatin isolation can also be performed by high salt extraction (Rodriguez-Collazo et al., 2009), it is not ideal for mass spectrometry if salt clean-up is not effective. High salts notoriously interfere with derivatization steps, form salt adducts and be detrimental to electrospray ionization. Therefore, we and others use a mild detergent lysis step followed by centrifugation to isolate nuclei, reducing sample handling and thereby loss (Das et al., 2009; Bhanu et al., 2019). While extraction of histones from eukaryotic cells is straightforward, organisms with the cell wall like yeast need harsher pretreatment with homogenization, alkali, sonication, hypotonic swelling and boiling (Kizer et al., 2006). After chromatin isolation, we then extract histones in acids like H2SO4 or HCl, in which they are highly soluble due to their high isoelectric point. H2SO4 is the most used acid; however, selected histones and histone PTMs are more soluble and stable in other acids. For example, extraction of H1 is more favorable in HCl, while labile PTMs like phosphorylations, differential extraction of isoforms as well as novel PTMs may benefit from high salt extraction in neutral pH.
The protocol we present here is focused for human myoblasts and myotubes, but it is easily translatable for most cell types. We describe a propionylation procedure to derivatize lysine side chains prior trypsin digestion. We also recommend the addition of a synthetic peptide for quality control (QC) of the sample preparation, and its products can be monitored for assessing over- and under-propionylation, and trypsin digestion (Sidoli et al., 2019).
Following derivatization and digestion, the peptides are desalted off-line in an easy-to-assemble, in-house stage tip. Because of the high hydrophilicity of the peptides generated with our protocol (Sidoli et al., 2016), we use a Porous Graphitic Carbon (PGC) resin on top of a C18 plug for desalting, thereby increasing the dynamic range of detection and quantification. Desalting can be performed using 96-well plates to multiplex the sample preparation. After eluting peptides from the desalting column, the samples are ready to spray. We recommend to utilize automated direct injection (DI-MS) methods like the TriVersa NanoMate (Advion), so samples can be injected in a robotized manner. We describe in the protocol the details to prepare a dedicated acquisition method to analyze the described histone peptides, including differential quantification of isobaric forms. In addition, we describe our software EpiProfile Lite (Sidoli et al., 2019) for automated signal extraction and relative quantification of histone PTMs. Finally, we give helpful indications for correlating PTM analysis to their epigenetic significance, so that follow-up studies can be designed by the investigators.
To sum up, we outline a standard improved one-pot workflow for ultra-rapid analysis of histone modifications with direct-injection mass spectrometry. In this protocol, we simplified the most laborious steps in sample preparation and allow for efficient sample preparation with a QC peptide. We demonstrate high-throughput analysis of 344 samples by multiplexed desalting and the DI-MS acquisition that allows for < 1 min runs into the mass spectrometer. Rapid data analysis was also possible due to about 2 MB RAW files generated by DI-MS, as against 1,300 MB file sizes from nLC/MS2 analyses. This workflow allows comprehensive systems level analysis yielding quantitative information on 220 histone peptides and their PTMs for each of the 344 samples, all within 2-3 days (Figure 1), making it particularly suitable for biomarker discovery and validation of epigenetic diseases.
Materials and Reagents
- Glass pasteur pipettes (Neta Scientific, catalog number: 7095B-9 )
- Microcentrifuge tube adapter (GL Sciences, catalog number: 501021514 )
- 3M EmporeTM Solid Phase Extraction Disks C18 (CDS Analytical, catalog number: 98-0604-0218-1 )
- Hypercarb® 30-40 µm Carbon 150-300 Å (Thermo Scientific, catalog number: 60106-402 )
- Fused silica capillary 75 µm ID x 363 µm OD (Molex (Polymicro), catalog number: TSP075375 )
- 1.7 ml microcentrifuge tubes, Posi-click (Denville, catalog number: C2170 )
- 15 and 50 ml conical centrifuge tubes (Falcon, catalog numbers: 352196 , 352070 )
- TipOne tips for P1000, 200, 20 and 10 pipettes (USA Scientific)
- MS sample vial, LaPhaPack, Snap, 12 x 32 mm (LEAP PAL Parts, catalog number: LAP.11190933 )
- MS sample vial Snap Cap, PE, Natural, Pre-Slit (LEAP PAL Parts, catalog number: LPP.011-03-8303 )
- Deep Well, 96-Well Microplate, 2.0 ml (Thomas Scientific, catalog number: 89237526 )
- 96-well plate, V-Bottom 600 µl (Axygen, catalog number: P-DW-500-C-S )
- 96-well flat bottom plate (Fisher Scientific, catalog number: 12565501 )
- Sealing Mat for 96 Well PCR Microplates (Axygen, catalog number: 14-222-024 )
- Reprosil-Pur 120 C18-AQ 3 µm, 3 g (ESI Source Solutions, r13.aq.0003)
- 10x Dulbecco’s PBS without Ca2+/Mg2+ (Mediatech, catalog number: MT21031CM )
- Cell dissociation buffer (Thermo Fisher, catalog number: 13151014 )
- NP-40 Alternative (Sigma-Millipore, catalog number: 492016 )
- Dithiothreitol (DTT) (Sigma-Millipore, catalog number: 111474 )
- AEBSF (Sigma-Millipore, catalog number: 101500 )
- Microcystin (Sigma-Millipore, catalog number: 475818 )
- Sodium butyrate (Sigma-Millipore, catalog number: 817500 )
- Sulfuric acid (H2SO4) (Fisher Scientific, catalog number: A484-212 )
- Trichloroacetic acid (Fisher Scientific, catalog number: A322-100 )
- Acetone (Fisher Scientific, catalog number: A18-500 ), store at 20 °C
- Hydrochloric acid (HCl) (Fisher Scientific, catalog number: A144S-500 )
- Ammonium bicarbonate (NH4HCO3) (Fisher Scientific, catalog number: A643-500 )
- Bovine Serum Albumin (BSA) (Sigma-Millipore, catalog number: A7906-100G )
- Coomassie Brilliant Blue G-250 (Fisher Scientific, catalog number: BP100-25 )
- Acetonitrile, Optima LC/MS grade (Fisher Scientific, catalog number: A955-1 )
- Propionic anhydride (Sigma-Millipore, catalog number: 8006080100 )
- Ammonium hydroxide (Fisher Scientific, catalog number: A669S-500 )
- Modified Trypsin, sequencing grade (Fisher Scientific, catalog number: PRV5113 )
- Glacial acetic acid (Fisher Scientific, catalog number: 18-602-907 )
- Trifluoroacetic acid, Optima LC/MS grade (Fisher Scientific, catalog number: A116-05AMP )
- Formic Acid, Optima LC/MS grade (Fisher Scientific, catalog number: A117-50 )
- Water, Optima LC/MS grade (Fisher Scientific, catalog number: W6-4 )
- Methanol, Optima LC/MS grade (Fisher Scientific, catalog number: A456-4 )
- Trypsin-digested BSA MS standard (CAM-modified) (New England Biolabs, catalog number: P8108S )
- Sodium chloride (NaCl) (Fisher Scientific, catalog number: BP358-1 )
- Potassium chloride (KCl) (Fisher Scientific, catalog number: AC424090250 )
- Magnesium chloride (MgCl2) (Fisher Scientific, catalog number: AC223211000 )
- Calcium chloride (CaCl2) (Fisher Scientific, catalog number: C79-500 )
- Nuclei isolation buffer (NIB-250) (see Recipes)
- NP-40 Alternative (see Recipes)
- Protease inhibitors (see Recipes)
- Phosphatase inhibitor (see Recipes)
- HDAC inhibitor (see Recipes)
- 0.2 M (0.4 N) sulfuric acid (see Recipes)
- 100% Trichloroacetic acid (TCA) (w/v) in ddH2O (see Recipes)
- Acetone + 0.1% hydrochloric acid (see Recipes)
- 100 mM Ammonium bicarbonate (see Recipes)
Caution: Avoid direct contact with reagents and research material; always use gloves and other appropriate personal protective equipment (PPE). Use automatic pipetting to prevent accidental ingestion.
Liquid nitrogen: Training required before use. Use face shield, cryogenic gloves, cryogenic apron and closed toed shoes while dispensing.
H2SO4, TCA, HCl and acetic acid are corrosive acids. Always open stock bottles of concentrated acid in a chemical fume hood to avoid inhalation of fumes; open dilute acid in a well-ventilated workspace.
Ammonium hydroxide is a strong base and should also be used in fume hood to avoid inhaling fumes.
Acetone, acetonitrile and methanol are flammable. Use precautions for fire hazard. Exercise standard exposure precautions. Store all acetone-containing reagents in -20 °C. It is also a strong organic solvent and will leach polystyrene pipettes. Use glass pipettes for dispensing.
Propionic anhydride is an irritant and is corrosive. Degrades in contact with atmospheric air and so it is stored under argon. Higher exposures are likely when topping with argon gas. Use face shield and goggles in addition to gloves and lab coat when topping. Note: This is DEA-controlled substance that needs approvals and not be available for prompt delivery.
Compressed gas (nitrogen, argon): Training required for proper securing and safe handling. Argon spray cans can also be used, instead of 14 L tanks.
Equipment
- -80 °C freezer
- Hypersep cartridge (Thermo Scientific, catalog number: 60109-404 )
- Ceramic scoring wafer (Restek, catalog number: 20116 )
- Pipetman, P1000, 200, 20 and 10 (Rainin)
- Cold centrifuge (700 x g-18,000 x g) (NuAire, model: Nuwind , catalog number: NU-C200V )
- Argon (Airgas, catalog number: AR HP 300 )
- Nitrogen (Airgas, catalog number: NI UHP300 )
- Dounce tissue grinder & pestle (Kimble Kontes, catalog number: 885300 )
- Mass spectrometer (Thermo Scientific, model: Orbitrap Fusion )
- Nanodrop (Thermo Scientific, model: ND3300 )
- pH indicator strips, Instachek (pH 0-13), Micro Essential Lab, model: Hydrion , catalog number: JR-113 )
- Pressure Injection Cell (Next Advance, model: PC77 )
- SpeedVac+ vacuum pump and plate rotor (Savant, model: SC210A )
- Tube rotator (Thermo Scientific, catalog number: 88881001 )
- Vortex Mixer (Thermo Scientific, catalog number: 88880017 )
- Triversa Nanomate (Advion, model: TR263 )
Software
- EpiProfile Lite (Sidoli et al., 2019). Available in GitHub at https://github.com/zfyuan/EpiProfileLite
Procedure
文章信息
版权信息
© 2020 The Authors; exclusive licensee Bio-protocol LLC.
如何引用
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Bhanu, N. V., Sidoli, S. and Garcia, B. A. (2020). A Workflow for Ultra-rapid Analysis of Histone Post-translational Modifications with Direct-injection Mass Spectrometry. Bio-protocol 10(18): e3756. DOI: 10.21769/BioProtoc.3756.
- Bhanu, N. V., Sidoli, S., Yuan, Z. F., Molden, R. C. and Garcia, B. A. (2019). Regulation of proline-directed kinases and the trans-histone code H3K9me3/H4K20me3 during human myogenesis. J Biol Chem 294(20): 8296-8308.
分类
生物化学 > 蛋白质 > 翻译后修饰
分子生物学 > 蛋白质 > 检测
您对这篇实验方法有问题吗?
在此处发布您的问题,我们将邀请本文作者来回答。同时,我们会将您的问题发布到Bio-protocol Exchange,以便寻求社区成员的帮助。