

Quantitative Kinetic Analyses of Histone Turnover Using Imaging and Flow Cytometry
组蛋白转换的定量动力学成像及流式细胞术研究
发布: 2020年09月05日第10卷第17期 DOI: 10.21769/BioProtoc.3738 浏览次数: 5479
评审: Imre GáspárYong-Yu LiuPooja Verma
参见作者原研究论文
The authors used this protocol in:

Jun 2019
Advertisement









Abstract
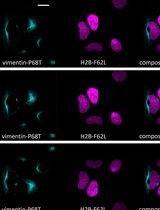
Dynamic histone changes occur as a central part of chromatin regulation. Deposition of histone variants and post-translational modifications of histones are strongly associated with properties of chromatin status. Characterizing the kinetics of histone variants allows important insights into transcription regulation, chromatin maintenance and other chromatin properties. Here we provide a protocol of quantitative and sensitive approaches to test the timing of incorporation and dissociation of histones using a two-color SNAP-labeling system, labelling pre-existing and newly-incorporated histones distinctly. Together with cell cycle synchronization methods and cell cycle markers, this approach enables a pulse-chase analysis to determine the turnover of histone variants during the cell cycle, detected using imaging or flow cytometry methods at single cell resolution. As well as testing global histone turnover, cell cycle-dependent cellular localization of histone variants can be also addressed using imaging approaches.
Keywords: Chromatin dynamics (染色质动力学)Background
Chromatin remodeling is part of the numerous fundamental cellular activities in eukaryotic cells (Geiman and Robertson, 2002; Clapier and Cairns, 2009). Accessibility by transcription factors and RNA polymerases are generally associated with changes in DNA methylation and chromatin states, including accessibility, post-translational histone modifications and deposition of histone variants. Histone variants differentially coordinate the gene expression that regulates development, cell differentiation or other physiological activities (Banaszynski et al., 2010). They also play diverse roles in DNA repair, telomere maintenance, heterochromatin formation and chromatin segregation (Henikoff and Smith, 2015; Zink and Hake, 2016). Moreover, dysregulation of a histone variant’s incorporation is associated with cancer (Vardabasso et al., 2014), indicating a significant role in human disease. To understand the kinetics of histone variants, including incorporation into and dissociation from specific chromatin regions during the cell cycle, is crucial since it tightly links regulatory properties with the mitotic maintenance of epigenetic (heritability from parent to daughter cells). DNA replication involves major chromatin remodeling to duplicate the entire chromatin structure following mitosis. Histones that associate with chromatin prior to DNA replication are transiently dissociated from DNA by the access of the DNA polymerase complex. Release of pre-existing histones randomly re-associate at newly synthesizing replication forks together with newly synthesized histones (Balhorn et al., 1975; Alabert and Groth, 2012; Annunziato, 2012). Pre-existing post-translational modifications on histones and some histone variants also re-associate on the newly synthesized DNA at the replication fork, explaining re-association of pre-existing histone at the replication fork is one part of the maintenance of mitotic inheritance of chromatin states. To determine the timing of post-translational modifications or incorporation of histone variants, a sensitive pulse-chase system that can distinguish the detection of newly incorporated histone from pre-existing histones is required. Newly synthesized canonical histones are unmodified when they are incorporated into chromatin during DNA replication. Unlike de novo DNA methylation, which occurs together with DNA replication (Tillo et al., 2016), most histone marks are not established on newly incorporated histones on the replicating fork. Proteomic based pulse-chase approaches (see in Alternative methods section below) have been used to determine the global kinetics of histone post-translational modifications by detecting the pre-existing and new deposition of histone acetylation and methylation at lysine residues (Pesavento et al., 2008; Scharf et al., 2009; Martinez-Garcia et al., 2011; Xu et al., 2011; Zee et al., 2012; Alabert et al., 2015). This approach identified two distinctive kinetic patterns of histone modifications. One group, such as histone H3 acetyl-lysine at 27 (H3 K27 ac), exhibits rapid turnover to equalize, and notably they are not maintained through the cell cycle (Scharf et al., 2009). This rapid acetylation kinetics probably represent temporally active transcription dynamics (Stasevich et al., 2014). While another group including H3 lysine tri-methylation at 9 (K9me3) or H3 K27me3 is acquired more slowly and step-wise from mono and di to tri-methylation, established during G1 phase following the cell cycle instead of before mitosis (Pesavento et al., 2008; Scharf et al., 2009; Martinez-Garcia et al., 2011; Xu et al., 2011; Zee et al., 2012; Alabert et al., 2015). Importantly, this group has the property of mitotic chromosome memory. The potential mechanism of maintenance of histone marks over cell division has been addressed by imaging approaches. In situ proximity ligation assays using specific antibodies against histone lysine methylations and their methyltransferases detected that histone modifiers continuously associate with the replicating DNA component in Drosophila embryos, suggesting the association of modifiers on replicating DNA may provide a “tag” to be methylated (Petruk et al., 2012).
Recent advances in chemical protein labeling technologies provide us with a powerful tool for protein tagging applications in living cells (e.g., SNAP (New England Biolabs), CLIP (New England Biolabs), Halo (Promega) and TMP (Active Motif) tag). These technologies are based on the covalent labeling of genetically encoded tags that bind with specific ligands conjugated to cell permeable substrates such as synthetic fluorescent dyes or biotin, which can mediate affinity purification in biochemical applications. In contrast with common genetically encoded tags, this chemical labeling of protein can be utilized in timing-dependent labeling with many choices of fluorophores, which allows the pulse-chase labeling of specific protein. This labeling technology has revealed the deposition timing of histone variants at specific chromatin architectures using imaging detection (e.g., CENP-A at centromeres [Dunleavy et al., 2009] and macroH2A at heterochromatin [Sato et al., 2019]).
Here, we provide a detailed protocol of a pulse-chase method using the SNAP-tag labeling system which has utilized quantitative histone variant detection with single cell resolution. Using this protocol, distinct histone kinetics, dissociation of pre-existing histones and association of newly synthesized histones, can be detected simultaneously. We describe two detection approaches, fluorescence microscopy and flow cytometry, as well as the detail of imaging analysis using FIJI/ImageJ software which is freely available (https://fiji.sc/).
Advantages and Limitations
The pulse-chase method using a chemical protein labeling system is an easy, non-hazardous and sensitive approach compared with a conventional pulse-chase approach using radioactive molecules (see in Alternative methods section). Most of the required reagents and fluorophores are commercially available. Global turnover of histones can be addressed using imaging and flow cytometric applications and notably, timing-specific localization at specific chromatin architectures also can be characterized with imaging approaches. Using this protocol, both pre-existing and newly incorporated histone variants can be detected simultaneously in the same cells with single cell resolution. Unlike radioisotope or metabolic labeling approaches, which detect endogenous histones, this labeling approach relies on the genetically encoded tags (e.g., SNAP-, CLIP-, Halo-tag) that can be linked with specific substrates. Therefore, a plasmid construct that expresses the target histones with SNAP-tag and its use in a stably expressing cell line are required. In addition, the localization and other biological functions of desired tagging histones must be examined to determine whether it remains functioning as an endogenous histone. Optimization of the construct (e.g., N-terminus or C-terminus tagging, changing the choice of promoter) and levels of expression might be necessary for obtaining accurate observations. Another limitation of this approach is that it is not applicable for detection of post-translational modifications of histones.
Alternative methods
Isotope labelling with proteomic detection
SILAC (Stable Isotope Labelling with Amino acids in Culture) followed by mass spectrometry is a powerful approach to investigate global turnover of endogenous histone variants and post-translational modifications (Yuan et al., 2014). In this approach, newly synthesized histones are labeled with radioactive heavy isotope and chase the turnover of labeled his tones compared with pre-existing histones containing light amino acids. Following mass spectrometry analysis determines the pre-existing and deposition of post-translational modifications or variants. This approach is suitable to detect global histone turnover, but is not able to detect histone marks at specific chromatin architecture or genomic loci.
Metabolic labeling with genome-wide approaches
Non-radioactive metabolic labeling of nascent proteins can be an alternative approach to label global newly synthesized histones. The approach, “Covalent Attachment of Tags to Capture Histones and Identify Turnover”, also called ‘CATCH-IT’, enables genome-wide investigation to characterize active histone replacement (Deal et al., 2010). This approach is based on the labeling scheme of nascent peptide by incorporation of methionine homolog, azidohomoalanine (Aha), which is generally used for the detection of active translation in cells. In this approach, the nucleosomes containing Aha-labeled newly synthesized histones are bioconjugated with biotin by a cycloaddition reaction (as known as “click” chemistry), and pulled down with streptavidin beads. Isolated DNA from pull-down was applied on a tiling microarray to determine the genomic loci that exhibit active histone replacement in Drosophila S2 cells. The characterized genomic sites that have active histone turnover correspond with the site of incorporation of histone H3 variant, H3.3, which is detected at transcriptionally active loci (Henikoff et al., 2009). This approach enables the detection of genomic loci with active turnover of histones.
Chemical labeling approaches such as the SNAP-tagging system can also be the alternative option to investigate genome-wide histone variant incorporation (Sato et al., 2019). In this approach, newly incorporated SNAP-tagged histones are linked with SNAP-biotin after the treatment of SNAP-Cell® Block (bromothenylpteridine, BTP), a non-fluorescent substrate to mask the reactivity of pre-existing histones. Then, biotin-linked, newly incorporated histones can be pulled down with streptavidin beads. The purified DNA fraction from pull-down samples can be sequenced with massive parallel sequencing. This approach might be useful to detect timing and genomic loci dependent incorporation of histone variants but unable to detect post-translational modifications.
Materials and Reagents
- 50-ml Falcon tubes
- 15-ml Falcon tubes
- 1.5-ml tubes
- Tissue culture plates (12 or 24-well; Falcon, catalog number: 353043 or 353047 )
- Tissue culture dishes (35 x 10 mm or 60 x 15 mm, and 100 x 20 mm, Falcon, catalog numbers: 353001 or 353002 and 353003 )
- Coverslips (18 or 12 mm No.1.5)
- Microscope slides (e.g., Fisher Scientific, catalog number: 12-544-2 )
- 5-ml FACS tubes (Fisher Scientific, catalog number: 149595 )
- Adherent cells of interest (e.g., mouse embryonic fibroblasts [MEFs], HEK293 cells)
- SNAP-Cell Oregon Green (New England BioLabs, catalog number: S9104S )
- SNAP-Cell Block (New England BioLabs, catalog number: S9106S )
- SNAP-Cell TMR-Star (New England BioLabs, catalog number: S9105S )
- Thymidine (Sigma, catalog number: T1895 )
- RO-3306 (Santa Cruz Biotechnology, catalog number: sc-358700 )
- Nocodazole (Sigma, catalog number: M1404 )
- Prolong Diamond Antifade Mountant (Thermo Fisher Scientific, catalog number: P36970 )
- Prolong Diamond Antifade Mountant with DAPI (Thermo Fisher Scientific, catalog number: P36962 )
- Drug for selection of stably expressing cells [e.g., 600-1,200 µg/ml G418 (geneticin), Zeocin (Thermo Fisher Scientific, catalog number: R25001 )]
- Click-iTTM EdU Cell Proliferation Kit for Imaging, Alexa FluorTM 647 dye (Thermo Fisher Scientific, catalog number: C10340 )
- pSNAPf Vector (New England BioLabs, catalog number: N9183S )
- pCCL-CellCycle (Sato et al., 2019)
- Suitable culture media and supplements [e.g., DMEM (Sigma, catalog number: D6429 ) supplemented with 10% FBS (Atlanta Biologicals, Inc., catalog number: S11150H ) and 100 U/ml Penicillin-Streptomycin (Sigma, catalog number: 15140148 )]
- EDTA, 0.5 M, pH 8.0, Molecular Biology Grade (Sigma, catalog number: 324506 )
- HEPES solution, 1 M, pH 7.0-7.6, sterile-filtered (Sigma, catalog number: H0887 )
- Coverslips coating solution, e.g., 0.01% Poly-L-lysine solution (Sigma, catalog number: P4707 ), collagen coating solution (according to the manufacturer's instruction, Sigma, catalog number: SAFC-125-50 )
- Sterile 1x PBS pH 7.4, no calcium, no magnesium for cell culture (e.g., Corning, catalog number: 21-031-CV )
- 10× DPBS, (e.g., Roche, catalog number: 11666789001 )
- 32% (wt/vol) paraformaldehyde (PFA) (Electron Microscopy Sciences, catalog number: 15714 )
!CAUTION: Paraformaldehyde is a hazardous solution and a cross-linking agent. Wear protective gloves and handle it under a fume hood. - Triton X-100, 0.1% (vol/vol) (Thermo Fisher Scientific, catalog number: BD 151500 )
- Nuclease-free water (Thermo Fisher Scientific, catalog number: 10977-015 )
- Immersion oil 1.518, for the microscope/objective
- Bovine serum albumin (BSA) (Sigma, catalog number: A7030 )
- Cloning of SNAP-histone expression vector and its stably expressing cell line (see Recipes)
- Fixation Buffer (see Recipes)
- Permeabilization buffer (see Recipes)
- Blocking buffer (see Recipes)
- Sorting Buffer (see Recipes)
Equipment
- Vortex mixer
- Tabletop centrifuge
- Pointed tip tweezer
- Water bath
- Biological hood/biosafety cabinet (for cell culture work)
- Cell culture incubator suitable for cell culture of your choice (e.g., 37 °C, 5% CO2)
- Wide-field fluorescent microscope (e.g., Olympus, model: BX-61 , equipped with four filter sets for DAPI (Semrock, model: DAPI-5060C-Zero ), Cy3 (Chroma, model: 41007 ), FITC (Semrock, model: FITC-5050A-Zero), and Cy5 (Semrock, model: Cy5-4040C-Zero ), an EXFO X-Cite Series 120 PC metal halide light source, Photometrics Cool SNAP HQ CCD camera, and molecular Devices Metamorph acquisition software or equivalent microscope set-ups or equivalent microscope system
- Flow cytometer (BD Biosciences, model: LSR II )
Software
- Metamorph software for image acquisition (https://www.moleculardevices.com/systems/metamorph-research-imaging/metamorph-microscopy-automation-and-image-analysis-software)
- ImageJ/FIJI (Schindelin et al., 2012) (avalable at https://fiji.sc/)
- FlowJo (https://www.flowjo.com/solutions/flowjo/downloads)
Procedure
文章信息
版权信息
© 2020 The Authors; exclusive licensee Bio-protocol LLC.
如何引用
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Sato, H., Singer, R. H. and Greally, J. M. (2020). Quantitative Kinetic Analyses of Histone Turnover Using Imaging and Flow Cytometry. Bio-protocol 10(17): e3738. DOI: 10.21769/BioProtoc.3738.
- Sato, H., Wu, B., Delahaye, F., Singer, R. H. and Greally, J. M. (2019). Retargeting of macroH2A following mitosis to cytogenetic-scale heterochromatic domains. J Cell Biol 218(6): 1810-1823.
分类
细胞生物学 > 细胞成像 > 荧光
细胞生物学 > 基于细胞的分析方法 > 流式细胞术
您对这篇实验方法有问题吗?
在此处发布您的问题,我们将邀请本文作者来回答。同时,我们会将您的问题发布到Bio-protocol Exchange,以便寻求社区成员的帮助。