

BRIDGE: An Open Platform for Reproducible Protein-Ligand Simulations and Free Energy of Binding Calculations
BRIDGE:一个可重复的蛋白质配体模拟和结合自由能计算的开放平台


(*contributed equally to this work) 发布: 2020年09月05日第10卷第17期 DOI: 10.21769/BioProtoc.3731 浏览次数: 8718
评审: Prashanth N SuravajhalaTianjiao HuangAnonymous reviewer(s)
参见作者原研究论文
The authors used this protocol in:

Sep 2019
Advertisement









Abstract
Protein-ligand binding prediction is central to the drug-discovery process. This often follows an analysis of genomics data for protein targets and then protein structure discovery. However, the complexity of performing reproducible protein conformational analysis and ligand binding calculations, using vetted methods and protocols can be a challenge. Here we show how Biomolecular Reaction and Interaction Dynamics Global Environment (BRIDGE), an open-source web-based compute and analytics platform for computational chemistry developed based on the Galaxy bioinformatics platform, makes protocol sharing seamless following genomics and proteomics. BRIDGE makes available tools and workflows to carry out protein molecular dynamics simulations and accurate free energy computations of protein-ligand binding. We illustrate the dynamics and simulation protocols for predicting protein-ligand binding affinities in silico on the T4 lysozyme system. This protocol is suitable for both novice and experienced practitioners. We show that with BRIDGE, protocols can be shared with collaborators or made publicly available, thus making simulation results and computations independently verifiable and reproducible.
Keywords: Protein Dynamics (蛋白质动力学)Background
The Googleplex of protein ligand binding possibilities is best reduced to a focussed set of drug hits, that can be interrogated in vitro and in vivo to produce a candidate drug for clinical studies, using computational chemistry methods. As a consequence, correct use of accurate compute methods that provide estimates of the energy of binding between a potential drug molecule, and a protein is central to the drug discovery process (Cournia et al., 2017). The idea is that the ligand should preferentially bind to the protein and inhibit its function. In this protocol, you will learn how to calculate relative protein-ligand binding free energies using open-source free energy tools and the Biomolecular Reaction and Interaction Dynamics Global Environment (BRIDGE) platform (Senapathi et al., 2019) that is built on the Galaxy platform (Afgan et al., 2018). The free energy differences between two ligands are calculated through an in silico perturbation from one ligand to another as they are i) bound to a protein (e.g., T4 lysozyme) and ii) solvated in water. Setting up free energy molecular simulations to run on High-Performance Computing (HPC) hardware resources, often done using scripts and the Linux command line, is challenging and a barrier to obtaining reliable results. In addition to the computational challenges, not all steps are repeatable as when run using varied software and hardware resources available through different laboratories, universities, public facilities, and cloud platforms. However, the objective of BRIDGE development is to provide a web-based platform that includes reliable methods for simulation, analysis, and analytics. A similar approach to BRIDGE is employed by the BioExcel Building Blocks software (Andrio et al., 2019). The BRIDGE-Galaxy platform makes the transition from genetics to proteomics to chemistry a seamless one and enables repeatable computer simulations and analysis using curated workflows.
We study T4 lysozyme as an illustration of a protocol to compute the relative binding of ligands, and we use benzene vs. p-xylene bound to T4 lysozyme as an example. The following tools are used: ProtoCaller for setup (Suruzhon et al., 2020), GROMACS for molecular dynamics (MD) simulations (Abraham et al., 2015), and alchemical analysis for simulation analysis (Klimovich et al., 2015). At least two simulations are required to compute the relative free energy of binding. First, the transformation between ligands in water and then the transformation between ligands bound to the solvated protein. Ligand 2 can be transformed into Ligand 1 or vice versa; the choice of direction is important to understand the meaning of the free energy calculated. Calculating in both directions is not required but can be used as an additional metric to confirm the computational convergence of the free energy result.
The identification of a protein target may be made through genomics experiments and bioinformatics analysis of gene expressions or arrived at through a literature search that may point to one or many protein targets. The protein structures are sourced from the RCSB Protein Data Bank. The identification of ligand hits is often guided by molecules that share a similarity to the known substrate or inhibitors found in the PDB, or derived from existing pre-screened molecule libraries, using similarity measure and docking calculations. Public molecular databases are good resources for finding possible ligands, for example, ChEMBL (Gaulton et al., 2012), ZINC (Irwin and Shoichet, 2005) and PubChem (Kim et al., 2016).
Preparing the protein from a PDB file into a state ready for simulation, posing the ligand in the binding site, and selecting appropriate parameters for accurate simulation are some of the immediate barriers to an MD simulation, even more so for free energy simulation. There are numerous alternative methods to prepare the protein, e.g., PDBFixer (Eastman et al., 2013). However, the preparation often requires specialist training to treat challenges such as missing loops from PDBs properly, repairing or installing mutated residues, disulfide bonds creation and assigning experimentally consistent protonation states. In the case of ligands, accurate charges require parameterization and assignment. In the protocol presented here, the automated setup of protein and ligand is done using the Alchemical Setup tool that links several specialized tools to perform protein setup and parameterization.
The BRIDGE interface (Figure 1) is straightforward and follows the Galaxy design. Tools are in the left panel, and a history of progress is in the right panel and information about the current tool or dataset selected is in the central panel. Data can be uploaded using the green upload icon at the top of the Tools menu (left panel), while the top menu bar includes your user account and other information. To search for tools, type a keyword into the search bar and tools with matching words will be displayed. Changes to the tools are versioned and the available versions can be selected via the central panel interface. Example histories of this process that include the outputs can be found by browsing to shared data and clicking on published histories (https://galaxy-compchem.ilifu.ac.za/histories/list_published). Choose the example history and click on the ‘+’ icon to add to your histories. Browse the example outputs using the eye icon. The free energy workflow illustrated in Figure 2 is also available (Reference 15).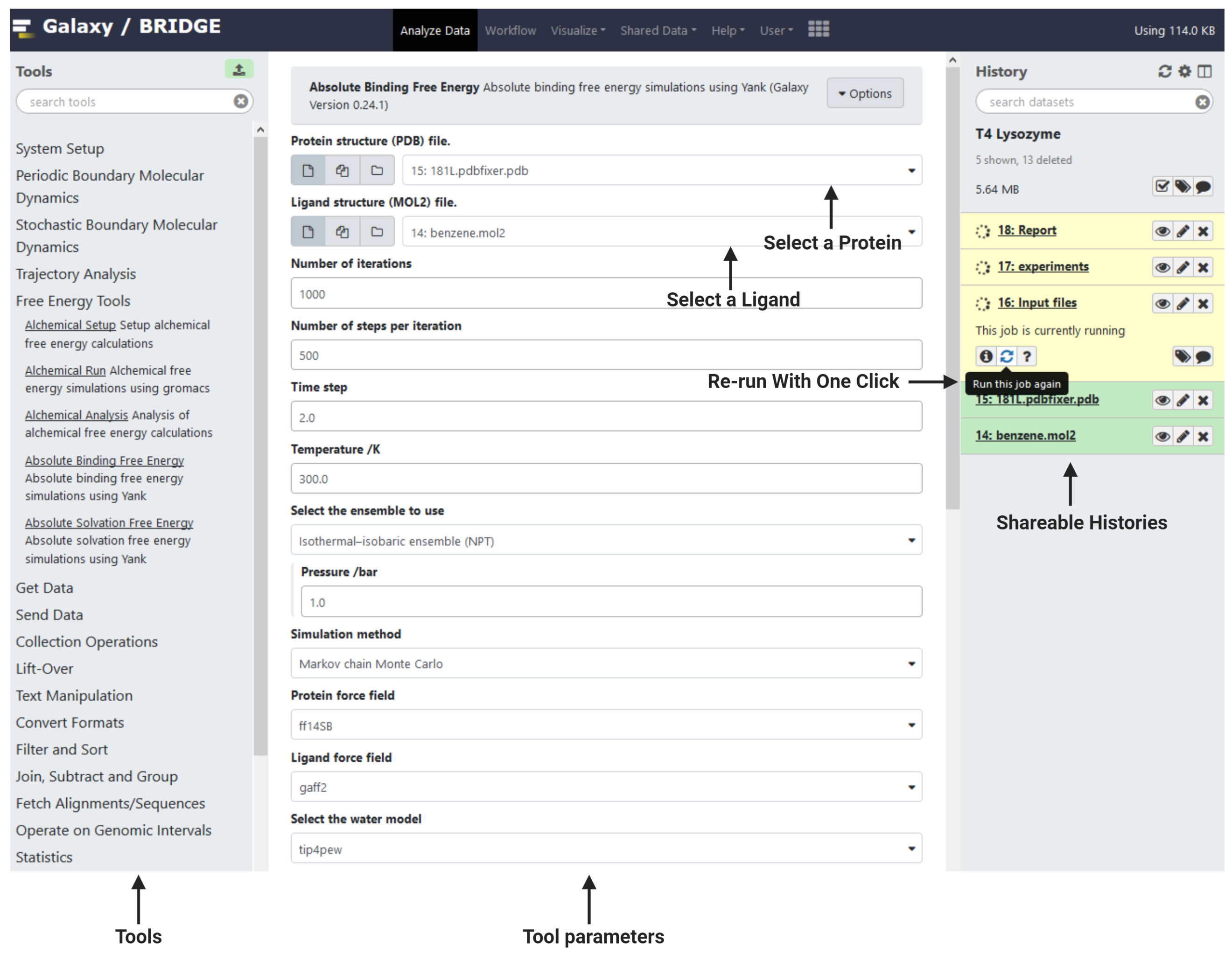
Figure 1. An overview of the Galaxy interface. Tools are in the left panel. Tool information or data are shown in the central panel and a history is displayed in the right panel.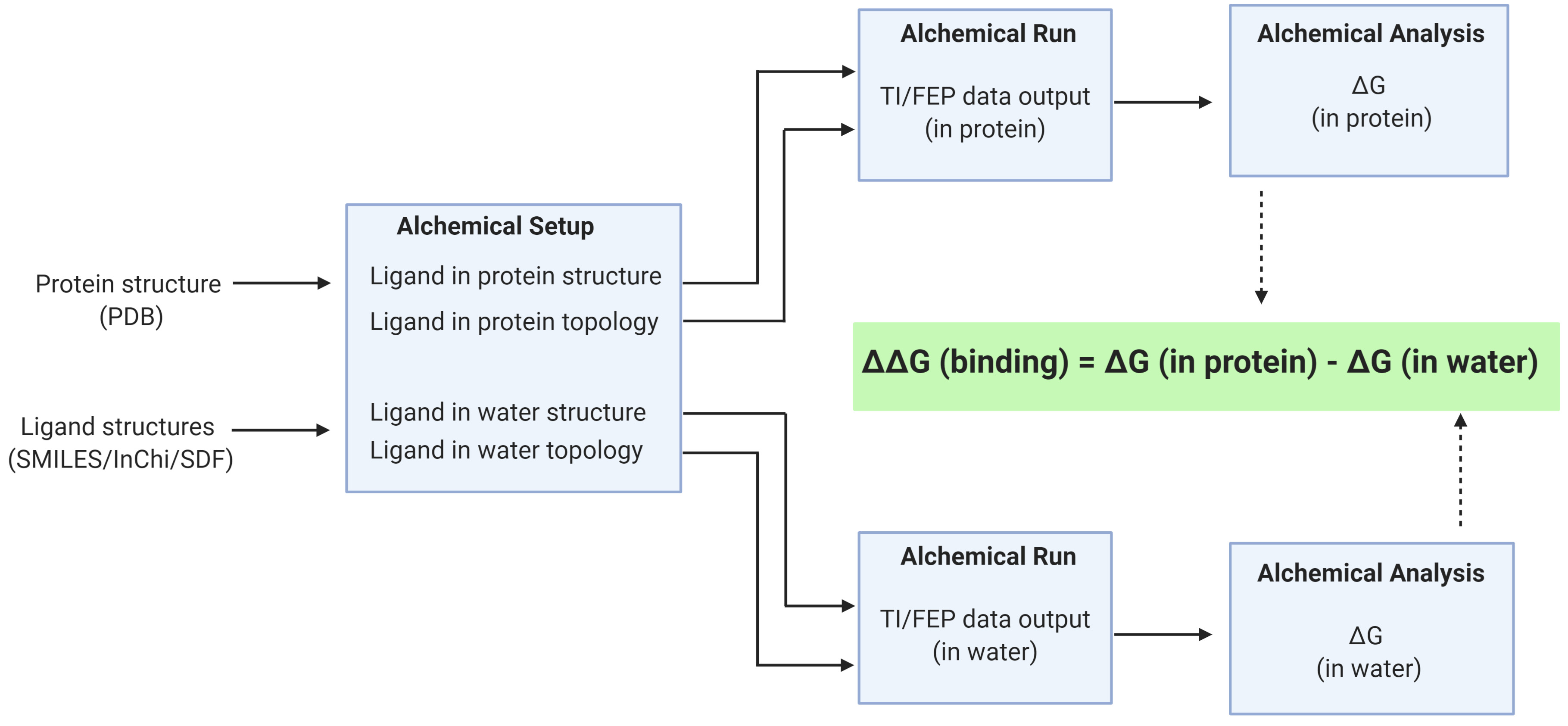
Figure 2. An overview of the Relative Binding Free Energy Workflow. The receptor in PDB format and ligand in SMILES are inputs to the Alchemical Setup. The multiple simulations are run using the Alchemical Run and the Alchemical Analysis tool is used to check convergence.
As part of the procedures below, a brief analysis is provided that is used to confirm that the free energy simulations are converged. An analysis of the molecular trajectories of the lead inhibitor can be conducted to identify the molecular interactions responsible for ligand binding and consider the conformation of ligand and protein. The aim of this further analysis would be to understand the rationale for binding. Further analysis might, for example, include hydrogen bond analysis and Ramachandran analysis, Root Mean Square Deviation (RMSD) and Principal Component Analysis (PCA). An example workflow is provided (Reference 14). PCA will reveal the flexibility of the catalytic domain and principal protein motions affecting inhibitor binding, while hydrogen bonding analysis would indicate key hydrogen bonding interactions between the ligand and the protein binding site.
Software
- Local computer
To run this on your local compute resources, obtain the BRIDGE Docker (https://github.com/scientificomputing/bridge-docker) and install the Docker image on your local machine. All libraries, compilers and links are packaged within the Docker image, so installation is seamless. A quad-core processor with 8GB RAM and 80GB hard drive space is sufficient for testing with Docker on any operating system. - Public servers
Access is obtained through a web browser and software installation is not required. The BRIDGE platform can be accessed at https://galaxy-compchem.ilifu.ac.za/. The tools are available on Galaxy Europe (http://cheminformatics.usegalaxy.eu).
Procedure
文章信息
版权信息
© 2020 The Authors; exclusive licensee Bio-protocol LLC.
如何引用
Senapathi, T., Barnett, C. B. and Naidoo, K. J. (2020). BRIDGE: An Open Platform for Reproducible Protein-Ligand Simulations and Free Energy of Binding Calculations. Bio-protocol 10(17): e3731. DOI: 10.21769/BioProtoc.3731.
分类
分子生物学 > 蛋白质 > 蛋白质-蛋白质相互作用
系统生物学 > 蛋白质组学 > 空间蛋白组学
您对这篇实验方法有问题吗?
在此处发布您的问题,我们将邀请本文作者来回答。同时,我们会将您的问题发布到Bio-protocol Exchange,以便寻求社区成员的帮助。