

High-throughput Flow Cytometry Assay to Investigate TDP43 Splicing Function
高通量流式细胞术检测TDP43剪接功能
发布: 2020年04月20日第10卷第8期 DOI: 10.21769/BioProtoc.3594 浏览次数: 5819
评审: Steven BoeynaemsBede PortzEmilie Besnard
参见作者原研究论文
The authors used this protocol in:

Oct 2019
Advertisement









Abstract
Mutations in RNA-binding proteins (RBPs) such as TDP43 are associated with transcriptome-wide splicing defects and cause severe neurodegenerative diseases, including amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD). The impact of RBP mutations on splicing function is routinely studied using PCR-based bulk measurements. However, the qualitative and low-throughput nature of this assay make quantitative and systematic analyses, as well as screening approaches, difficult to implement. To overcome this hurdle, we have developed a quantitative, high-throughput flow cytometry assay to investigate TDP43 splicing function on a single-cell level
Background
RNA-binding proteins (RBPs) such as TDP43 regulate post-transcriptional gene regulation by orchestrating RNA stability, transport and processing, including mRNA splicing (Gerstberger et al., 2014). Impairment of RBP function as a result of mutations and/or aggregation has been implicated in the etiology of many neurodegenerative diseases such as amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) (Harrison and Shorter, 2017). Notably, ALS-associated mutations in RBPs like TDP43 can cause transcriptome-wide splicing defects, suggesting that misregulation of RBP splicing function may be a key driver of disease (Arnold et al., 2013; Sun et al., 2015). Thus, an important goal in ALS research is to uncover the molecular mechanisms of RBP function, requiring experimental approaches for the systematic interrogation of RBP splicing function.
Widely-used methods for studying splicing are reverse transcription-PCR (RT-PCR) and RNA sequencing (RNA-seq). Both methods can be used to study the splicing of endogenous transcripts; however, typically only as a bulk measurement of an entire population of cells. RT-PCR is simple and affordable, yet efficient only to study splicing of a select few endogenous transcripts. Moreover, RT-PCR read-outs are often only semi-quantitative. In contrast, RNA-seq provides quantitative insights into global splicing, but is time- and cost-intensive and requires sophisticated data analysis pipelines (Wang et al., 2009). Thus, both RT-PCR and RNA-seq are poorly suited for high-throughput applications, for example to systematically compare the splicing function of multiple RBP variants.
A powerful tool to study splicing are minigenes, which typically are plasmid-based, simplified genes containing introns and exons that recapitulate defined splicing events outside of the native gene context (Baralle and Baralle, 2005). In particular, minigenes have been instrumental in discovering cis- and trans-acting splicing elements (Cooper, 2005). Instead of endogenous targets, minigenes are also routinely used to streamline investigation of RBP splicing function (D'Ambrogio et al., 2009; Kino et al., 2011). Conventionally, the splicing of minigenes is evaluated with analytical or quantitative RT-PCR. However, the low-throughput and bulk read-out of this method precludes leveraging the full potential of minigene-based splicing reporters.
To overcome these hurdles, we developed a minigene that encodes for a fluorescent protein (FP minigene), thus allowing us to convert a defined splicing event into a sensitive, inherently quantitative and readily detectable signal on a single-cell level by flow cytometry (Schmidt et al., 2019). To normalize splicing of the FP minigene to the overall abundance of the parent transcript, we fused it to another, intron-free fluorescent protein (constitutive FP). While this approach is broadly applicable (Gurskaya et al., 2012; Sorenson and Stevens, 2014; Gonatopoulos-Pournatzis et al., 2018), we adapted it specifically to study TDP43-dependent splicing by interrupting the FP minigene with exon 9 of the CFTR gene, which is skipped only in the presence of functional TDP43 (Buratti et al., 2001). To rapidly compare the splicing efficiency of multiple TDP43 variants in TARDBP (i.e., the gene encoding TDP43) knock-out cells, we used a bidirectional promoter to drive the coordinated expression of both our splicing reporter and a given BFP-tagged TDP43 variant from the same plasmid. A graphical overview of our splicing reporter design in shown in Figure 1.
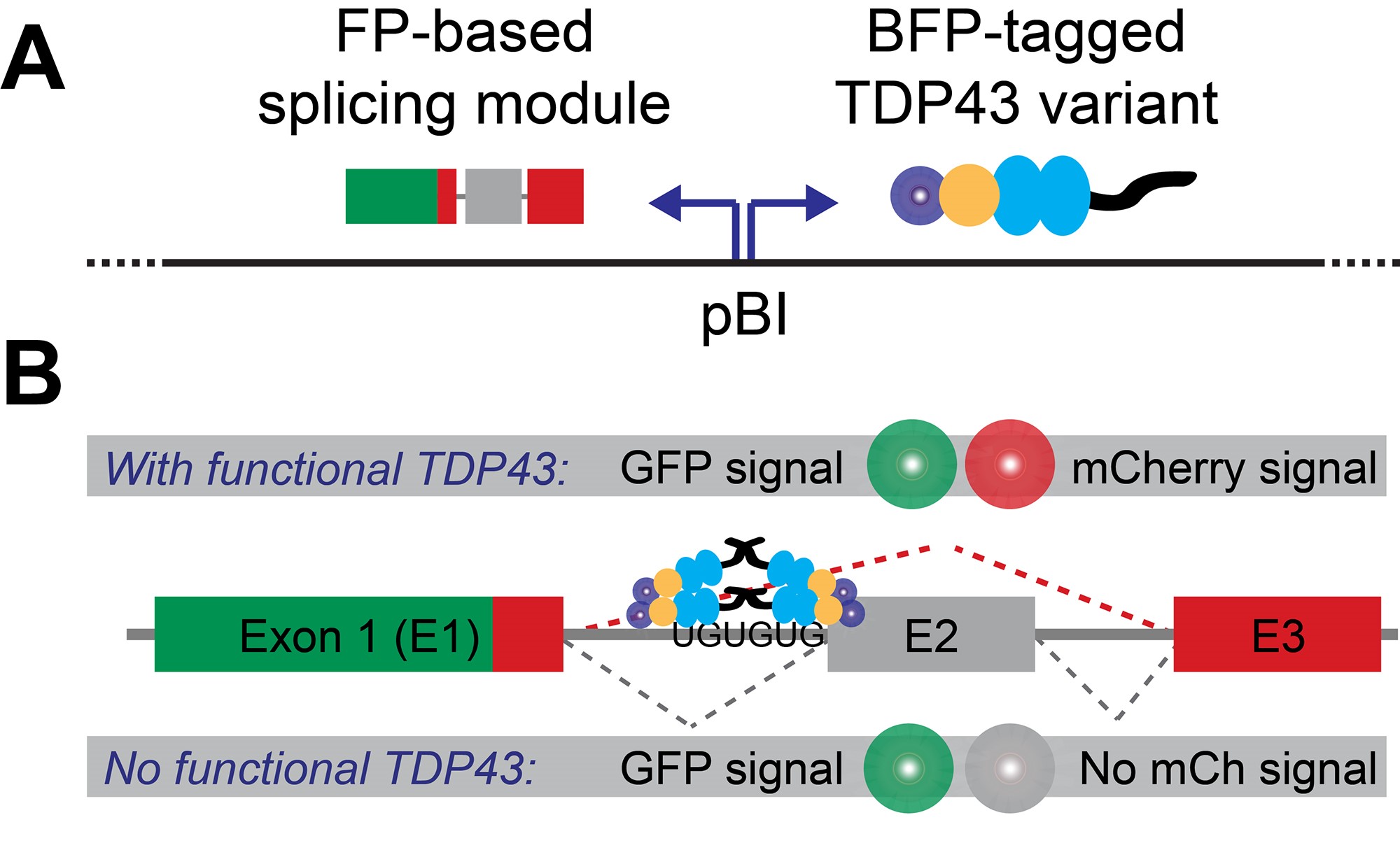
Figure 1. Splicing reporter design. A. The fluorescent protein (FP)-based splicing module and BFP-tagged TDP43 variants are expressed from the same plasmid via a bi-directional promoter (pBI). B. Production of a dually-fluorescent GFP-mCherry fusion protein depends on the presence of functional TDP43, which is required to mediate the skipping of CFTR gene-derived exon (labeled E2).
Here, we describe in detail how to perform and analyze our flow cytometry-based splicing assay. This protocol is optimized for use with HEK-293T wild-type and HEK-293T TARDBP knock-out cells (Schmidt et al., 2019), yet in principle suitable for other recently published constitutive or inducible TARDBP knock-out cell lines as well (Chiang et al., 2010; Gasset-Rosa et al., 2019; Roczniak-Ferguson and Ferguson, 2019).
Materials and Reagents
- Cell lines
- HEK-293T wild-type cells (ATCC, catalog number: CRL-3216 )
- HEK-293T TARDBP knock-out cells (available upon request)
- Cell culture
- 1.5 ml microtubes (Eppendorf, catalog number: 0 22363204 )
- 60 µm scepter tips for cell counting (Milipore-Sigma, catalog number: PHCC60050 )
- 10 cm tissue culture plates for propagating cell lines (Thermo Scientific, catalog number: 12-556-002 )
- 6-well tissue culture plates for splicing assays (Thermo Scientific, catalog number: 14-832-11 )
- 70 µm cell strainer (Corning, Falcon, catalog number: 08-771-2 )
- 5 ml round-bottom polystyrene tubes (Corning, Falcon, catalog number: 14-959-1A )
- SH800S flow cytometer setup beads (Sony Biotechnology, catalog number: LE-B3001 )
- HyClone DMEM high glucose (GE Healthcare, catalog number: SH30081FS ) supplemented with:
- 10% FBS (Atlanta Biologicals, catalog number: S11150H )
- 1 mM sodium pyruvate (Gibco, catalog number: 11-360-070 )
- 2 mM L-glutamine (Gemini Biosciences, catalog number: 400106 )
- 1x MEM non-essential amino acids (Gibco, catalog number: 11-140-076 )
- 40 U/ml penicillin and 40 µg/ml streptomycin (Gemini Biosciences, catalog number: 400109 )
- Sterile 1x PBS at pH 7.4 (Sigma-Aldrich, catalog number: P3813-5x10PAK )
- Trypsin-EDTA (Gemini Biosciences, catalog number: 400150 )
- X-tremeGENE9 transfection reagent (Roche, catalog number: 0 6365787001 )
- OptiMEM (Gibco, catalog number: 31985-070 )
- Plasmids (available at https://www.addgene.org/Rajat_Rohatgi/, unless otherwise noted)
- Splicing reporter only: pHBS1389 IBB-GFP-mCherry3E (Addgene, catalog number: 118803 )
- Positive control: pHBS1503 [IBB-GFP-mCherry3E]-[BFP-TDP43 WT] (Addgene, catalog number: 133327 )
- Negative control: pHBS1501 [IBB-GFP-mCherry3E]-[BFP-TDP43 ∆RRM] (Addgene, catalog number: 133325 )
- Extensive collection of TDP43 CTD mutants in the splicing reporter (e.g., Addgene, catalog numbers: 133328-133333 )
- Backbone for subcloning of CTD mutants into splicing reporter: pHBS1224 [IBB-GFP-mCherry3E]-[BFP-TDP43 ∆CTD EcoRV] (available upon request)
- Compensation probes: mammalian expression vectors with BFP, GFP and mCherry inserts (e.g., Addgene, catalog numbers: 54665 , 54759 and 54563 )
Equipment
- Single-channel pipettes (Rainin or equivalent)
- Cell culture incubator (Thermo Scientific or equivalent)
- Scepter cell counter (Milipore-Sigma) or equivalent manual/automatic cell counter
- Sony SH800S cell sorter (Sony Biotechnology) or equivalent cell sorter equipped with 405 nm, 488 nm and 561 nm lasers, as well as appropriate filters
- Computer for data analysis (e.g., 3.1 GHz Intel i7 processor with 16 GB RAM)
Software
- SH800S operating software for data collection (Sony Biotechnology) or operating software for equivalent flow cytometers
- Mathematica Version 11.3 or higher (Wolfram Research) + custom data analysis pipelines:
- Script ‘CSA_Gating.nb’ (available at https://github.com/RohatgiLab/TDP43-analysis)
- Script ‘CSA_Plotting.nb’ (available at https://github.com/RohatgiLab/TDP43-analysis)
Procedure
文章信息
版权信息
© 2020 The Authors; exclusive licensee Bio-protocol LLC.
如何引用
Schmidt, H. B. and Rohatgi, R. (2020). High-throughput Flow Cytometry Assay to Investigate TDP43 Splicing Function. Bio-protocol 10(8): e3594. DOI: 10.21769/BioProtoc.3594.
分类
分子生物学 > RNA > RNA 剪接
神经科学 > 神经系统疾病 > 神经退行性病变
细胞生物学 > 基于细胞的分析方法 > 流式细胞术
您对这篇实验方法有问题吗?
在此处发布您的问题,我们将邀请本文作者来回答。同时,我们会将您的问题发布到Bio-protocol Exchange,以便寻求社区成员的帮助。