

Detection of Heteroplasmic Variants in the Mitochondrial Genome through Massive Parallel Sequencing
大规模平行测序检测线粒体基因组异质粒

(*contributed equally to this work) 发布: 2019年07月05日第9卷第13期 DOI: 10.21769/BioProtoc.3283 浏览次数: 5525
评审: Chiara AmbrogioEnrico PatruccoMauro Sbroggio'
参见作者原研究论文
The authors used this protocol in:

Jul 2018
Advertisement









Abstract
Detecting heteroplasmies in the mitochondrial DNA (mtDNA) has been a challenge for many years. In the past, Sanger sequencing was the main option to perform this analysis, however, this method could not detect low frequency heteroplasmies. Massive Parallel Sequencing (MPS) provides the opportunity to study the mtDNA in depth, but a controlled pipeline is necessary to reliably retrieve and quantify the low frequency variants. It has been shown that differences in methods can significantly affect the number and frequency of the retrieved variants.
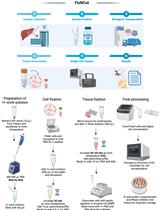
In this protocol, we present a method involving both wet lab and bioinformatics that allows identifying and quantifying single nucleotide variants in the full mtDNA sequence, down to a heteroplasmic load of 1.5%. For this, we set up a PCR-based amplification of the mtDNA, followed by MPS using Illumina chemistry, and variant calling with two different algorithms, mtDNA server and Mutect.
The PCR amplification is used to enrich the mitochondrial fraction, while the bioinformatic processing with two algorithms is used to discriminate the true heteroplasmies from background noise. The protocol described here allows for deep sequencing of the mitochondrial DNA in bulk DNA samples as well as single cells (both large cells such as human oocytes, and small-sized single cells such as human embryonic stem cells) with minor modifications to the protocol.
Background
In the past, the methods used for studying the mtDNA were amongst others PCR-RFLP (PCR-restriction fragment length polymorphism), Sanger sequencing and mitochondrial DNA re-sequencing using Affymetrix’s MitoChip. However, these methods are not able to accurately quantify the heteroplasmic load under 10%. Massive Parallel Sequencing (MPS) represents, in all likelihood, the best solution to investigate variants in the mitochondrial genome. However, when analyzing mtDNA there are two key factors that make mtDNA analysis less straightforward compared to the nuclear genome. First, the mtDNA contains regions with significant homology to regions dispersed within the nuclear genome called nuclear mitochondrial DNA sequences (NuMTS). Second, multiple mtDNA copies are present within a cell, and variants can be present at frequencies ranging between 0 and 100%. For this reason, when performing MPS analysis of the mtDNA different strategies might be applied to accurately identify and quantify single nucleotide variants (SNVs). The best approach to overcome the first problem is to enrich the sample for its mitochondrial genome only. This can be achieved by selectively amplifying the mtDNA by long-range PCR, by isolating the mtDNA using mtDNA enrichment kits, or by isolating the mitochondria themselves prior to DNA extraction (Just et al., 2015). The second issue is more challenging. Whilst MPS provides the ideal type of data to simultaneously identify SNVs and/or rearrangements and calculating their loads, the manner in which the data are generated and processed will not only determine the type of variants that can be detected, but also their lower threshold. If an SNV is present at a very low frequency, its signal will be undistinguishable from the systems sequencing errors (Bai and Wong, 2004; Rohlin et al., 2009; Zhang et al., 2012; Ye et al., 2014). Recently, many pipelines have been released to identify and quantify variants. However, bioinformatic processing does not represent the only critical step in these analyses. In our hands, we found that the initial amplification of the template is an extremely important step for the correct evaluation of SNV frequencies, such that a suboptimal PCR amplification leads to a gross overestimation of the retrieved frequencies (Zambelli et al., 2017). This is especially the case for PCR protocols with higher cycle numbers and primer sets that do not result in a linear amplification. The method we present here can be used to accurately detect and quantify single nucleotide variants at a low heteroplasmic load (as low as 1.5%) in both bulk DNA samples and single cells (Zambelli et al., 2017 and 2018). We here describe amplification conditions and bioinformatic processing for both bulk DNA and single cells with detailed information and screenshots about the bioinformatic steps.
Materials and Reagents
- 96-well plate
- 200 μl Eppendorf tubes
- LongAmp Buffer (LongAmp Taq DNA Polymerase kit) (New England Biolabs, catalog number: M0323L), stored at -20 °C
- Taq LongAmp (LongAmp Taq DNA Polymerase kit) (New England Biolabs, catalog number: M0323L), stored and kept at all times at -20 °C
- dNTP’s (dNTP sets, IllustraTM) (VWR, catalog number: 28-4065-57), diluted to 2 mM and stored at -20 °C
- Tricine (Sigma-Aldrich, catalog number: T9784), diluted to 200 mM and stored at 4 °C
- DTT (DL-Dithiothreitol) (Sigma, catalog number: D-0632), stored at 4 °C
- NaOH (Stock solution: 1 M)
- TBE buffer (1x) (Thermo Fisher Scientific, InvitrogenTM, catalog number: 15581044)
- Agarose (Agarose DNA Pure Grade, VWR, catalog number: 443666A)
- PCR Purification Kit (AMPure XP for PCR purification, Beckman Coulter, catalog number: A63882)
- Primer set 1 (5042f-1424r) (Integrated DNA Technologies, diluted to 10 μmol, aliquoted and stored at -20 °C)
Forward primer: 5′-AGC AGT TCT ACC GTA CAA CC-3′
Reverse primer: 5′-ATC CAC CTT CGA CCC TTA AG-3′ - Primer set 2 (528f-5789r) (Integrated DNA Technologies, diluted to 10 μmol, aliquoted and stored at -20 °C)
Forward primer: 5′-TGC TAA CCC CAT ACC CCG AAC C-3′
Reverse primer: 5′-AAG AAG CAG CTT CAA ACC TGC C-3′
Note: These two primer sets (Items 12 and 13) were selected because they were able to amplify large fragments of the mtDNA and tested negative when amplifying Rho Zero cells, indicating that they did not amplify NuMTS in the nuclear genome. These primer sets were also evaluated for their performance in low frequency heteroplasmy calling by performing spike-in experiments and were shown to give the better estimation of the low frequency variants (Zambelli et al., 2017).
- QubitTM dsDNA Broad Range Assay Kit (InvitrogenTM, catalog number: Q32853)
- Ethanol (80%)
- Tris-HCl solution (10 mM, pH 8.0)
- Library Preparation kit (KAPA HyperPlus kit, Roche, catalog number: 07962436001)
- Fragmentation Kit (HS NGS Fragment Kit, Agilent, catalog number: DNF-474)
- Tween 20 (Sigma-Aldrich, catalog number: P9416)
- MPS Reagent Kit (NovaSeq6000 S2 Reagent Kit [200 cycles]) (Illumina, catalog number: 20012861)
- Gel electrophoresis (see Recipes)
- Alkaline Lysis Buffer (see Recipes)
- EBT (Elution Buffer with Tris) buffer (see Recipes)
Equipment
- Agarose Gel electrophoresis apparatus
- Power Supply (Electrophoresis Power Supply–EPS 301) (GE Health Care, catalog number: 18113001)
- Thermal Cycler (VeritiTM 96-Well, Applied Biosystems, catalog number: 4375786)
- Magnet
- Qubit Fluorometric Quantification (InvitrogenTM, catalog number: Q33238)
- Fragment Analyzer (Agilent formerly Advanced Analytical Technologies, Agilent, https://www.aati-us.com/instruments/fragment-analyzer/)
- Sequencing system (e.g., NovaSeq6000, Illumina, catalog number: 20012850)
Software
- bcl2fastq conversion software v2.19 script (Illumina, http://emea.support.illumina.com/downloads/bcl2fastq-conversion-software-v2-19.html)
- Seqtk (https://bioconda.github.io/recipes/seqtk/README.html)
- BWA-MEM
- GATK v3.3 (Broad Institute, https://software.broadinstitute.org/gatk/)
- GATK v3.6 Mutect2 (Broad Institute, https://software.broadinstitute.org/gatk/documentation/tooldocs/3.7-0/org_broadinstitute_gatk_tools_walkers_cancer_m2_MuTect2.php)
- Mpileup (SAMtools, http://samtools.sourceforge.net/mpileup.shtml)
- mtDNA server (https://mtdna-server.uibk.ac.at/index.html)
- MitoWheel (http://mitowheel.org/mitowheel.html)
- MitImpact (http://mitimpact.css-mendel.it/)
- MutPred2 (http://mutpred.mutdb.org/)
Procedure
文章信息
版权信息
© 2019 The Authors; exclusive licensee Bio-protocol LLC.
如何引用
Mertens, J., Zambelli, F., Daneels, D., Caljon, B., Sermon, K. and Spits, C. (2019). Detection of Heteroplasmic Variants in the Mitochondrial Genome through Massive Parallel Sequencing. Bio-protocol 9(13): e3283. DOI: 10.21769/BioProtoc.3283.
分类
分子生物学 > DNA > DNA 测序
您对这篇实验方法有问题吗?
在此处发布您的问题,我们将邀请本文作者来回答。同时,我们会将您的问题发布到Bio-protocol Exchange,以便寻求社区成员的帮助。