

Rapid Multiplexed Reduced Representation Bisulfite Sequencing Library Prep (rRRBS)
快速多重简并代表性亚硫酸氢盐测序文库的制备
发布: 2019年02月20日第9卷第4期 DOI: 10.21769/BioProtoc.3171 浏览次数: 7035
评审: Shan JiangAjit ShahAnonymous reviewer(s)
参见作者原研究论文
The authors used this protocol in:

Oct 2018
Advertisement












Abstract
DNA methylation is a common mechanism of epigenetic regulation involved in transcriptional modulation and genome stability. With the evolution of next-generation sequencing technologies, establishing quantitative genome-wide DNA methylation profiles is becoming routine in many laboratories. However, many of these approaches take several days to accomplish and use subjective PCR methods to amplify sequencing libraries, which can induce amplification bias. Here we propose a rapid Reduced Representation Bisulfite Sequencing (rRRBS) protocol to minimize PCR amplification bias and reduce total time of multiplexed library construction. In this modified approach, the precise quantification of the final library amplification step is accomplished and monitored by qPCR, instead of using standard PCR and gel electrophoresis, to determine the appropriate number of cycles to perform. The main advantages of this rRRBS method are: i) Reduced amount of amplification enzyme used for library prep, ii) Reduced number of PCR cycles resulting in less PCR amplification bias, and iii) Preparation of quality multiplexed rRRBS libraries in only ~2 days.
Keywords: DNA methylation (DNA甲基化)Background
With the development of high-throughput sequencing technologies, evaluation of genome-wide DNA methylation profiles is more accessible than before. For the human genome, various commercial targeted methyl capture sequencing panels and methylation arrays are available and allow high-throughput, quantitative interrogation of methylation sites at single-nucleotide resolution. However, most of these commercial tools are not offered for other model species. Thus, cost-effective sequencing-based methods for genome-wide methylation analysis, not targeting species-specific genomic sequences, are still highly relevant and valuable especially when profiling large cohorts of samples. One of these techniques is Reduced Representation Bisulfite Sequencing (RRBS). This method enables you to quantitatively investigate the DNA methylation profile of ~10% of the overall CpGs, clustered in fragments composing approximately 1% of the genome, with a preference for CpG rich regions (e.g., CpG islands, promoters) (Meissner et al., 2005 and 2008; Smith et al., 2009; Gu et al., 2011).
Here, we present a rapid RRBS (rRRBS) protocol (Piché et al., 2018) that significantly decreases hands-on time, allowing to perform the entire protocol in ~2 days compared to the 7 days usually needed (Gu et al., 2011; Boyle et al., 2012). Importantly, by using a qPCR amplification step, we now minimize amplification bias during multiplexed library construction (Figure 1). Also, compared to the original and other RRBS protocols (Gu et al., 2011; Boyle et al., 2012; McGraw et al., 2015) our modified protocol reduces the quantity of reagent and enzyme used as we no longer test for the optimal number of cycles needed before the final library amplification step. This method is suitable for DNA methylation studies from cells or tissues from any common research species (e.g., human, mouse, rat, zebrafish).
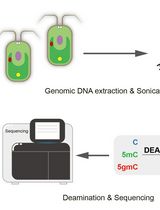
Figure 1. Graphical summary of the principal step of the rRRBS procedure
Materials and Reagents
- Pipette tips
- 1.5 ml DNase/RNase-free microtubes
- 0.2 ml DNase/RNase-free PCR strip tubes with individual caps
- QIAamp DNA micro or mini kit (QIAGEN, catalog numbers: 56304 or 51306)
- QuBit dsDNA BR assay kit (Thermo Fisher Scientific, catalog number: Q32853)
- Unmethylated λ phage DNA (Promega, catalog number: D1521)
- MspI 100 U/μl (New England Biolabs, catalog number: R0106M)
- Klenow fragment 5,000 U/ml (New England Biolabs, catalog number: M0212L)
- dNTP set (4 x 100 mM) (Thermo Fisher Scientific, catalog number: 10297018)
- AMPure XP magnetic beads in PEG solution (Beckman Coulter, catalog number: A63881)
- NEBNext® Multiplex Oligos for Illumina® (Methylated Adapter, Index Primers Set 1) (New England Biolabs, catalog number: E7535L)
- T4 DNA ligase 2,000,000 U/ml (New England Biolabs, catalog number: M0202M)
- EpiTect Fast bisulfite kit (QIAGEN, catalog number: 59826)
- SYBR Green, 10,000x (Thermo Fisher Scientific, Invitrogen, catalog number: S7563)
- KAPA HiFi Uracil+ 2x mastermix (Roche, catalog number: KK2802)
- Bioanalyzer High Sensitivity DNA Assay (Agilent, catalog number: 5067-4626)
- DNase/RNase free water
- EB buffer (QIAGEN, catalog number: 19086)
- NEB Buffer 2 (New England Biolabs, catalog number: B7002S)
- Ethanol 100%
Equipment
- P10, P20 pipets
- QuBit fluorometric device (Thermo Fisher Scientific, catalog number: Q33226)
- Vortex
- Mini-centrifuge
- Thermocycler
- Heating block
- DynaMagTM-96 Side Magnet (Thermo Fisher Scientific, catalog number: 12331D)
- DynaMagTM-2 Magnet (Thermo Fisher Scientific, catalog number: 12321D)
- qPCR machine
- qPCR plates2100
- Bioanalyzer instrument (Agilent, catalog number: G2939BA)
- Illumina sequencing apparatus
- -20 °C freezer
Procedure
文章信息
版权信息
© 2019 The Authors; exclusive licensee Bio-protocol LLC.
如何引用
Legault, L., Chan, D. and McGraw, S. (2019). Rapid Multiplexed Reduced Representation Bisulfite Sequencing Library Prep (rRRBS). Bio-protocol 9(4): e3171. DOI: 10.21769/BioProtoc.3171.
分类
分子生物学 > DNA > DNA 测序
您对这篇实验方法有问题吗?
在此处发布您的问题,我们将邀请本文作者来回答。同时,我们会将您的问题发布到Bio-protocol Exchange,以便寻求社区成员的帮助。