

往期刊物2013
卷册: 3, 期号: 20
癌症生物学

Soft Agar Anchorage-independent Assay
软琼脂锚着非依赖性分析

Xenograft Tumor Growth Assay
异种移植肿瘤增生分析

Protocol for T-cell Adhesion Strength on Tumor Cells under Flow Conditions
流动状态下T淋巴细胞对肿瘤细胞的粘着强度分析



细胞生物学

Bimolecular Fluorescence Complementation (BiFC) Assay for Direct Visualization of Protein-Protein Interaction in vivo
通过双分子荧光互补(BiFC)试验进行体内蛋白质之间的相互作用的可视化分析


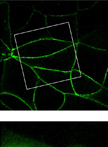
Measurement of Junctional Protein Dynamics Using Fluorescence Recovery After Photobleaching (FRAP)
采用荧光漂白恢复法(FRAP)测定接合蛋白质的动力学


Fluorescence Recovery After Photobleaching (FRAP) in the Fission Yeast Nucleus
裂殖酵母细胞核的荧光光漂白技术(FRAP)


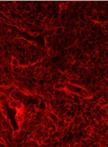
Hypertrophy Analysis and Quantification in Embryonic Cardiomyocites
胚胎心肌细胞的肥大分析和量化

Isolation of Whole Mononuclear Cells from Peripheral Blood
从外周血液中分离全单核细胞

Colon Tissue Immunoelectron Microscopy
结肠组织免疫电镜分析

免疫学
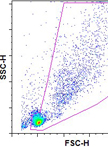
In vitro T Cell–DC and T Cell–T Cell Clustering Assays
T淋巴细胞与树突细胞以及T淋巴细胞之间的体外簇集试验
微生物学
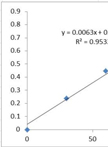
Surface Polysaccharide Extraction and Quantification
表面多糖提取和量化


Cell Fractionation and Quantitative Analysis of HIV-1 Reverse Transcription in Target Cells
靶细胞中HIV-1 逆转录的细胞分级分离和量化分析


Immunoblot Analysis of Histone H4 Acetylation and Histone H2A Phosphorylation in Candida albicans
白色念珠菌中组蛋白H4乙酰化和组蛋白H2A磷酸化的免疫印迹分析


神经科学
![[3H]-螺旋哌丁苯与多巴胺D2、D3 和D4受体的饱和结合](https://en-cdn.bio-protocol.org/imageup/arcimg/945.jpg?t=1774085190)
[3H]-Spiperone Saturation Binding to Dopamine D2, D3 and D4 Receptors
[3H]-螺旋哌丁苯与多巴胺D2、D3 和D4受体的饱和结合
![[3H]-螺旋哌丁苯与多巴胺D2、D3和 D4受体的竞争性结合](https://en-cdn.bio-protocol.org/imageup/arcimg/944.jpg?t=1774085190)
[3H]-Spiperone Competition Binding to Dopamine D2, D3 and D4 Receptors
[3H]-螺旋哌丁苯与多巴胺D2、D3和 D4受体的竞争性结合
植物科学
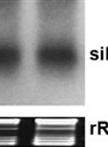
A High Resolution Short Interfering RNA (siRNA) Detection Method from Virus-infected Plants
高通量检测病毒感染植株中siRNA



Trehalase Activity in Arabidopsis thaliana Optimized for 96-well Plates
一种适用于利用96孔板测定海藻糖酶活性的方法


干细胞

Immunolabelling of Thin Slices of Mouse Descending Colon and Jejunum
小鼠降结肠和空肠薄切片的免疫标记
