- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

High-throughput Method for Determination of Seed Paternity by Microsatellite Markers

Published: Vol 3, Iss 8, Apr 20, 2013 DOI: 10.21769/BioProtoc.452 Views: 13496
Original research article
The authors used this protocol in:

Aug 2012
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
In this protocol, determination of seed paternity by microsatellite markers in Nicotiana attenuata is described. However, this does not include a protocol for the novel marker selection/identification, but rather exploits the markers generated for a closely related species N. tabacum (Bindler et al., 2007). This is a high-throughput protocol optimized and streamlined for one skilled person to process 384 (96 x 4) seeds in 5 days, from DNA isolation (from seedlings) to paternity assessment by microsatellite genotype data.
Keywords: Microsatellite genotypingMaterials and Reagents
- Agencourt Chloropure Kit (this kit is discontinued by Beckman Coulter but can be ordered) or Quiagen MagAttract 96 DNA plant core kit (QIAGEN, catalog number: 67163 )
- Qiagen Type-it Multiplex PCR kit (QIAGEN, catalog number: 206243 )
- QIAquick PCR purification kit (QIAGEN, catalog number: 28106 )
- Liquid nitrogen
- 100% Isopropanol, ultrapure
- Freshly prepared 70% ethanol made with nuclease free water
Note: 70% ethanol is hygroscopic, always make fresh for optimal result. - Agarose
- Ethydium bromide solution
- Nuclease free water
- Pure molecular biology grade ethanol (96– 100%)
Note: Calculate the volume of ethanol needed before you start. A new kit of 24 plates needs about 5 L of ethanol. - GENSCAN 500 ROX (Applied Biosystems, catalog number: 401734 )
- 1x TAE (Tris-Acetate-EDTA) buffer (see Recipes)
Equipment
- 2.2 ml Ritter Deep-well plates (ABGene, catalog number: AB-6661 )
- Adhesive plate film (ABGene, catalog number: AB-0558 , AB-626 , AB-662 )
- 4 mm steel balls (SPEX SamplePrep, catalog number: 2150 )
- Pipette tips
- Processsing Plate: Deep-well titer plate with a 96-well format (SPEX SamplePrep, catalog number: 2210 )
- Destination Plate: 300 μl round bottom microtiter plate (96 well, 300 μl well capacity, round bottom) (Corning, Costar®, catalog number: 07-200-105 )
- Desktop centrifuge for 96 well plates
- Reagent reservoir
- Genogrinder (SPEX SamplePrep, catalog number: 2010 )
- Cooling block (SPEX SamplePrep, catalog number: 2665 )
- Steel ball dispenser (SPEX SamplePrep, catalog number: 2100 )
- Multi-channel pipettes (0.1-10, 10-100, 30-300 μl)
- Agencourt Supermagnet magnetic Plate (Agencourt SPRIPlate 96R-Ring Magnet Plate, catalog number: A29164 )
- BioRad Gel casting tray, running tray, power pack etc (Bio-Rad Laboratories)
- Nanodrop spectrophotometer (Nanodrop)
- Pipette (0.2-2 μl), pipette tips, soft tissue paper
- ABI 3100 Genetic Analyzer (Applied Biosysyems)
Procedure
- Sample grinding (Day 1)
- Take up to 40 mg fresh tissue sample (leaf punches/seedling) in 2.2 ml plate. Take care to group similar samples/treatments/experimental designs together for easier data handling during sequencing and genotype analysis.
- Dispense 2 steel balls in each well with dispenser.
- Balance two plates exactly for grinding in Genogrinder.
- Seal the plate(s) with adhesive plate film.
- Cool the plate(s) in liquid nitrogen.
- Cool the cooling racks along with the plates.
- Grind in Genogrinder at Speed 250, 1x, for 1 min. If not crushed completely, again for 1 min.
- Take up to 40 mg fresh tissue sample (leaf punches/seedling) in 2.2 ml plate. Take care to group similar samples/treatments/experimental designs together for easier data handling during sequencing and genotype analysis.
- DNA isolation (Day 1)
Assembly in step B-1 is performed once for each new Agencourt Chloropure kit. If you have already made the following preparations for a previous experiment, please skip ahead to step B-2. Standard RNase treatment can be included either at the step B-5 with the homogenized lysate (requires large amount of enzyme, but convenient for high-throughput) or selectively after determination of the degree of RNA contamination at "quality check".- Add 80 ml of 100% Isopropanol to the wash buffer bottle provided with the kit. After addition of Isopropanol invert the bottle to mix. Once the solution has been thoroughly mixed, store at room temperature.
- Prepare bind buffer: Combine 6 μl bind buffer with 150 μl of 100% isopropanol for each individual isolation in a nuclease free vessel of suitable size (for example: for 10 isolations, add 60 μl of bind buffer to 1.5 ml of 100% isopropanol in a 15 ml conical tube). Vortex bind buffer bottle thoroughly before combining. Unused combined solution should be discarded.
- Homogenize each sample in 300 μl of lysis buffer, provided with the kit. (sample input should not exceed three 6 mm lyophilized leaf punches or 40 mg ground seeds or fresh leaf material.)
- Centrifuge lysate for 10 min at 1,100 RCF at room temp to pellet debris.
- Transfer 150 μl of homogenized lysate to 1.2 ml processing plate.
- Pipette 150 μl bind buffer (prepared in step B-2), mix by slowly pipetting 5 times and incubate at room temperature for 5 min.
- Move the plate onto the Agencourt Supermagnet and separate for 2-4 min. Wait for the solution to clear before proceeding to the next step.
- Slowly aspirate the cleared solution from the plate and discard.
Note: Aspirate from the top down to avoid disturbing the pellet. This step must be performed while the plate is situated on the magnet. Do not disturb the separated magnetic beads. If beads are drawn out, leave a few microliters of supernatant behind. - Remove the plate from the magnet and add 300 μl of Wash buffer. Pipette mix 10 times and incubate for 1 min at RT.
- Return plate to the magnet and separate for 2-4 min. Wait for the solution to clear before proceeding to the next step.
- Slowly aspirate the cleared solution from the plate and discard. This step must be performed while the plate is situated on the magnet.
- Remove the plate from the magnet and add 300 μl of 70% ethanol. Pipette mix 10 times to re-suspend the beads.
- Return plate to the magnet and separate for 2 min. Wait for the solution to clear before proceeding to the next step.
- Slowly aspirate the cleared solution from the plate and discard.
- Repeat steps l-n for a total of 2 ethanol washes.
- Let the plate air dry for 5 min at room temperature. The plate should air-dry until the last visible traces of ethanol evaporate. Over-drying the sample may result in a lower recovery.
- Remove the plate from the magnet and add 50 μl of nuclease-free water. Re-suspend the beads by pipette mixing 10 times.
Note: Smaller or larger elution volumes can be used for more or less concentrated product; however the minimum elution volume should be 40 μl to ensure complete elution. Optimal elution volumes need to be experimentally determined.- DNA quality/quantity check
1) Quality
a) Prepare 0.8% agarose gel with 1x TAE buffer. Add 2 μl Ethydium Bromide before casting.
b) Load 2 μl DNA+1 μl loading dye
c) Add 1 kb or 100 bp ladder for reference
d) Run at constant 100 V for 30 min
e) Check DNA for quality, presence of RNA and quantity in gel doc system and record. - Dilution of DNA sample
a) Dilute the samples to 50 ng/μl as working stock.
b) Dispense them in 3 PCR plates (for 3 multiplex PCR reactions) similarly and maintaining the sample organization of working DNA plate. Make a record of the samples.

Figure 1. DNA quality check on 0.8% agarose gel. Lane 1-6, 8-13 represent extracted DNA samples, lane 7 represents DNA marker.
2) Quantitya) Follow the procedure to measure concentration of DNA in Nanodrop. b) Export to excel file for reference. - DNA quality/quantity check
- Add 80 ml of 100% Isopropanol to the wash buffer bottle provided with the kit. After addition of Isopropanol invert the bottle to mix. Once the solution has been thoroughly mixed, store at room temperature.
- PCR amplification (Day 2)
- Multiplex groups
Primer groups are selected based on primer sequences using software MultiPLX version 2.0 (available at http://bioinfo.ut.ee/?page_id=167, Ref: doi: 10.1093/bioinformatics/bti219) to select multiplex grouping. Medium stringency was chosen to select groups.
Example of 3 multiplex groups of 8 primer pairs:
Table 1. Example of 3 multiplex groups of 8 primer pairs:
- 10x Primer mix
- First make 100 μM primer stock (if using 50 μM primer stock, adjust the volume of the table below accordingly).
- Mix all primers according to the table below to get 3 primer-mix groups (sufficient for 1x PCR plate i.e. 96 samples).
Table 2. Schematic representation of preparation of 3 primer mix groups:
- First make 100 μM primer stock (if using 50 μM primer stock, adjust the volume of the table below accordingly).
- PCR reaction mix
- Thaw the 2x Type-it Multiplex PCR Master Mix (MM) (if stored at -20 °C), template DNA (already dispensed in PCR plates), RNase-free water, and the primer mix. Mix the solutions completely before use.
Note: It is important to mix the solutions completely before use to avoid localized concentrations of salts. - Prepare a reaction mix according to the table below. Note: The reaction mix typically contains all the components required for multiplex PCR except the template DNA. Prepare a volume of reaction mix 10% greater than that required for the total number of reactions to be performed. For reaction volumes less than 25 μl, the 1:1 ratio of Type-it Multiplex PCR Master Mix to primer mix and template should be maintained.
Note: Starting with an initial Mg2+ concentration of 3 mM is recommended as provided by the 2x Type-it Multiplex PCR MM.
Table 3. Reaction Mix preparation for one 96 well plate samples
(modified after manufacturer's instruction)
- Mix the reaction mix thoroughly and dispense appropriate volumes into PCR tubes or plates. Note: Mix gently, for example, by pipetting the reaction mix up and down a few times. Due to the hot start, it is not necessary to keep samples on ice during reaction setup.
- Thaw the 2x Type-it Multiplex PCR Master Mix (MM) (if stored at -20 °C), template DNA (already dispensed in PCR plates), RNase-free water, and the primer mix. Mix the solutions completely before use.
- PCR condition
Table 4. PCR condition (according to manufacturer's instruction)
- PCR programme:
Ctrl. Tube
Lid- 105 °C
(1)95 °C-05:00 min
(2)95 °C-00:30 min
(3)60°C- 01:30 min
(4)72 °C-00:30 min
(5)Go to step (2), repeat 29
(6)60°C-30:00 min
(7)20 °C- ∞
(8)End
(9)Store PCR products in -20 °C, till further processing. - PCR check
Check PCR products randomly on 1.8% agarose gel (this is just to check if everything is working before proceeding to next steps. So choose random samples). Load 4 μl product and 100 bp ladder.
Note: Use only Xylene Cyanol loading dye as Bromophenol Blue dye interfere with the amplified band range.
- Multiplex groups
- PCR purification (Day 3)
- Add 75 μl nuclease free water to each well of PCR plate (containing 25 μl PCR product) to make it 100 μl.
- In a 96 well plate (at-least 1 ml capacity) dispense 300 μl Buffer PM. Add 100 μl PCR sample and mix by pipetteing.
- Apply the samples to the wells of the QIAquick plate while it is fitted with the collecting deepwell plate.
- Centrifuge at 1,100 RCF (g) for 2 min.
- Discard flow-through.
Note: If the sample volume is more than 600 μl apply the remaining samples to the wells of the QIAquick plate and repeat step 4. - Wash wells of QIAquick plate by adding 900 μl of Buffer PE to each well and centrifuge at 1,100 RCF (g) for 2 min.
- Repeat step 4-f.
- Discard flow-through and gently soak any buffer remaining on the tip of column with soft tissue.
- Centrifuge at 1,500 RCF (g) for 4 min.
Important: This step removes residual Buffer PE from the membrane. - Let the plate dry for 5 min or until all the ethanol (of Buffer PE) evaporates. Place it under fume hood for 10 min is sufficient.
- To elute, place the plate on collection tubes (provided), add 50 μl of RNase-free water (provided) to the center of each well of the QIAquick 96 plate, incubate for 2 min, and centrifuge at 1,500 RCF (g) for 2 min.
Important: Ensure that the elution buffer is dispensed directly onto the center of QIAquick membrane for complete elution of bound DNA. Please note that the average eluate volume is 60 μl from 80 μl elution buffer volume, and 40 μl from 60 μl elution buffer volume.- Quality check
Check Purified PCR product by nanodrop or 1.8% agarose gel.
Note: For nanodrop analysis, centrifuge the samples at 1,500 RCF for 2 min and estimate from the upper layer (to precipitate any remaining silica particles from the column, which sometimes interfere with sensitive nanodrop estimation). Imporant: It is better to check randomly the purified product on 1.8% agarose gel to be sure of quality. As this is a multiplex PCR, do not expect a clear band. The gel will appear as smear of many bands.
Figure 2. PCR amplification check on 1.8% agarose gel. Lanes 1-15 represent PCR products (with a couple of failed amplification on lane 7 and 14), lane 16 represents 100bp DNA marker. - Dilution
It is not needed to further dilute the samples if you start with 50 ng DNA sample and followed the procedure exactly. At this step the concentration of samples should be around 250 ng/μl.
For the size sequencing set up, it is recommended to dilute samples in this step to facilitate sample handling.
Add 200 μl nuclease free water to each purified sample tube (1/5th dilution). So the concentration becomes 50 ng/μl.
- Quality check
- Add 75 μl nuclease free water to each well of PCR plate (containing 25 μl PCR product) to make it 100 μl.
- Size sequencing (Day 4)
- Sequencing mix
The size sequence mix for each sample contains- 5 μl purified PCR product (250 ng total)
- 4.5 μl nuclease free water
- 0.5 μl GENSCAN 500 ROX marker
For a single sequence plate (prepare for 105 samples), set up MM as below:
- Mix 472.5 μl nuclease free water with 52.5 μl of ROX marker.
- Dispense 5 μl of above MM to each of sequencing plate.
- Mix in 5 μl purified PCR samples.
- Do not leave any sample well blank. Fill it with Nuclease free water.
- Cover the plate with adhesive seal mat.
- Centrifuge the plates briefly at 1,500 RCF.
- 5 μl purified PCR product (250 ng total)
- Prepare sequencing table of samples.
Prepare the sample list conveniently for recognition later. e.g. Sample name_primer mix group_Sample number_any other info.
Table 5. Sample list example
- Sequencing mix
- Analysis (Day 5)
Before starting analysis, import the data files generated by the sequencer to the computer with GeneMapper software. It is recommended to read the manual if you want to modify and fine-tune the marker settings and allele size binning for genotyping.- Genotype table
- Genotyping in GeneMapper
1) Go to 'file' menu and create new project. Name it conveniently.
2) Import your samples that belong to one primer mix group.
3) Create marker panel and bin set for each multiplex primer groups.
Refer to the manual for modifying or creating a new marker panel for new set of markers/primer mix in microsatellite. This involves:a) Marker set creation
b) Autobinning of alleles
c) Manual binning of alleles
d) Create new marker panel and bin set.
4) Use predefined marker panel for respective samples- Go to 'file' menu and create new project. Name it conveniently.
- Import your samples that belong to one primer mix group.
- Create marker panel and bin set for each multiplex primer groups
- Refer to the manual for modifying or creating a new marker panel for new set of markers/primer mix in microsatellite. This involves:
- Marker set creation
- Autobinning of alleles
- Manual binning of alleles
- Create new marker panel and bin set.
- Marker set creation
- Use predefined marker panel for respective samples
- Go to 'file' menu and create new project. Name it conveniently.
- Analysis parameters
1) Select the table setting as ‘Microsatellite default’
2) Select the analysis method as ‘Microsatellite analysis method’
3) Set the predefined panel as EV1 for primer mix 1 and so on.
4) Set size standard as GEN500.
5) Analyze
6) If some samples fail to match size standards, go to edit size standards and override size. If it does not improve the sizing, then there was some problem with the sample.
a) Open the genotype Tab. Do not bother about the GQ (genotype quality) column as it is set to a very high stringency.
b) Go to tools-panel manager-microsatellite kit-attenuata panel-EV1 binset. Select ‘show project alleles’. If you see any ‘*’ out of the grey bars (defines the range of allele bins), then either modify the range of existing bins (click on the grey bar and you’ll get handles for modification), or add a new bin (click on the blank space near your odd allele (that is not binned) and name it. Save the panel. Again analyze the samples with the modified binset.
c) If you want to see data for only two alleles and want to modify the way you want your data, modify the table output style. However, manual inspection of allele size, height etc is sometimes necessary to ensure optimal result than automation.
d) Go to file and export the genotype table in .csv or other excel compatible format.
- Genotyping in GeneMapper
- Paternity analysis in COLONY
For paternity/maternity analysis, i.e. to determine the parent of an offspring the software COLONY (http://www.zsl.org/science/research/software/colony,1154,AR.html).
COLONY is a Fortran program written by Jinliang Wang . It implements a maximum likelihood method to assign sibship and parentage jointly, using individual multilocus genotypes at a number of codominant or dominant marker loci. (Jones and Wang, 2010).- Formatting of genotype table.
1) Keep only the sample name, two allele sizes for each marker for all the marker.
2) Compile data for all the marker sets into a single datasheet for all offspring and all marker. - Marker table (save as tab delimited .txt file and rename 'marker').
Table 6. Marker details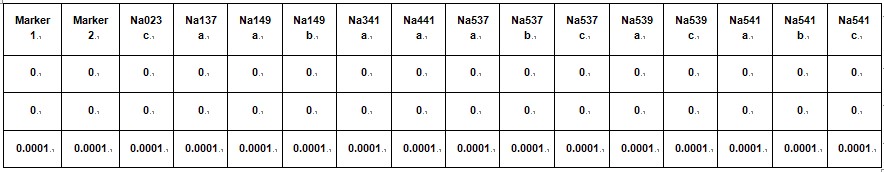
- Parent table (save as tab delimited .txt file and rename 'father/mother')
Table 7. Paternal allele list
Table 8. Maternal allele list
- Offspring table (save as tab delimited .txt file and rename 'offspring')
Table 9. Offspring allele list
- Analysis parameters
Feed all the information step by step as asked by the software and brose for the files to upload.
A typical parameter is as follows:
Number of loci:16
Number of offspring in the sample:
Number of male candidates:0
Number of female candidates:
Number of known paternal sibships:0
Number of known maternal sibships:0
Number of offspring with excluded fathers:0
Number of offspring with excluded mothers:0
Male mating system:Polygamy/monogamy
Female mating system:Polygamy/monogamy
Number of threads : 1
Number of Excluded Paternal Sibships:0
Number of Excluded Maternal Sibships:0
Seed for random number generator:1,234
Allele frequency:No updating by accounting for the inferred relationship
Species:Diploid/haploid
Sibship size prior:No
Known population allele frequency:No
Number of run:1
Length of run : short/medium/long
Monitor intermiediate results by:Every 1 second
Prob. a mum is included in the female candidates:0.5
Project data input produced:
NOTE to the Project: - Analysis outputs
1) Select Show result-'best configuration'. 2) Copy the table of assigned father ID/mother ID to each sample. 3) Sort parents and count offsprings sired by them in excel.
Population analysis in GenAlEx
- Formatting of genotype table.
- Population analysis in GenAlEx
When comparing populations for genotypes, or simply want to look at the genotypic diversity within a population, apart from parental analysis, it is better to use the excel addon 'GenAlEx' (http://biology.anu.edu.au/GenAlEx/Welcome.html). (Ref: Peakall R. and Smouse P.E. 2006. Genalex 6: Genetic analysis in Excel. Population genetic software for teaching and research, Molecular Ecology Notes, Volume 6, Issue 1, pages 288–295).
This can analyze:- Heterozygosity, F-statistics and Polymorphism
- Allelic Patterns
- Allele List
- Allele Frequencies and Principal Component Analysis (PCA).
- Heterozygosity, F-statistics and Polymorphism
- Genotype table
Recipes
- 1x TAE (Tris-Acetate-EDTA) buffer
- Prepare a stock solution of EDTA (ethylenediamine tetraacetic acid)
For a 500 ml stock solution of 0.5 M EDTA, dissolve 93.05 g EDTA disodium salt (FW = 372.2) in 400 ml deionized water and adjust the pH with NaOH to 8.0. Make up the final volume to 500 ml. - Prepare a 50x Stock Solution of TAE
Dissolve 242 g Tris base (FW = 121.14) in approximately 750 ml deionized water. Add 57.1 ml glacial acid and 100 ml of 0.5 M EDTA (pH 8.0) and adjust the solution to a final volume of 1 L. This stock solution can be stored at room temperature for a long time. The pH of this buffer needs no further re-adjustment and remains about 8.5. - Prepare a Working Solution of 1x TAE
Dilute the 50X stock solution by 1:50 with deionized water. Final solute concentrations are 40 mM Tris acetate and 1mM EDTA.
- Prepare a stock solution of EDTA (ethylenediamine tetraacetic acid)
Acknowledgments
This work was supported by the Max Planck Gesellschaft. The protocol was adapted from the publication: Kessler et al. (2012).
References
- Bindler, G., van der Hoeven, R., Gunduz, I., Plieske, J., Ganal, M., Rossi, L., Gadani, F. and Donini, P. (2007). A microsatellite marker based linkage map of tobacco. Theor Appl Genet 114(2): 341-349.
- Jones, O. R. and Wang, J. (2010). COLONY: a program for parentage and sibship inference from multilocus genotype data. Mol Ecol Resour 10(3): 551-555.
- Kessler, D., Bhattacharya, S., Diezel, C., Rothe, E., Gase, K., Schöttner, M. and Baldwin, I. T. (2012) Unpredictability of nectar nicotine promotes outcrossing by hummingbirds in Nicotiana attenuata. Plant J 71(4): 529-538.
Article Information
Copyright
© 2013 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Bhattacharya, S. and Baldwin, I. T. (2013). High-throughput Method for Determination of Seed Paternity by Microsatellite Markers. Bio-protocol 3(8): e452. DOI: 10.21769/BioProtoc.452.
Category
Molecular Biology > DNA > Genotyping
Plant Science > Plant molecular biology > DNA > Genotyping
Plant Science > Plant physiology > Tissue analysis
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.