- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Infection of Caenorhabditis elegans with Vesicular Stomatitis Virus via Microinjection
Published: Vol 7, Iss 22, Nov 20, 2017 DOI: 10.21769/BioProtoc.2617 Views: 9317
Reviewed by: Peichuan ZhangSuprabhat MukherjeeAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Mar 2017
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
Over the past 15 years, the free-living nematode, Caenorhabditis elegans has become an important model system for exploring eukaryotic innate immunity to bacterial and fungal pathogens. More recently, infection models using either natural or non-natural nematode viruses have also been established in C. elegans. These models offer new opportunities to use the nematode to understand eukaryotic antiviral defense mechanisms. Here we report protocols for the infection of C. elegans with a non-natural viral pathogen, vesicular stomatitis virus (VSV) through microinjection. We also describe how recombinant VSV strains encoding fluorescent or luciferase reporter genes can be used in conjunction with simple fluorescence-, survival-, and luminescence-based assays to identify host genetic backgrounds with differential susceptibilities to virus infection.
Keywords: Vesicular stomatitis virusBackground
Given its genetic tractability, small size, inexpensive culture, and transparent body, the free-living nematode Caenorhabditis elegans offers many advantages as a model organism. Furthermore, the susceptibility of C. elegans to a wide range of human bacterial and fungal pathogens has made the worm an attractive system for studying microbial pathogenesis (Zhang and Hou, 2013; Cohen and Troemel, 2015). More recently, the discovery of the positive-sense ssRNA Orsay virus (OV) as the first natural viral pathogen of C. elegans has prompted the use of the OV-C. elegans model to define nematode antiviral defense mechanisms (Felix et al., 2011; Gammon, 2017). These studies have demonstrated a clear role for nematode antiviral RNA interference pathway components, such as Dicer-related helicase 1 (DRH-1), in the restriction of virus replication (Ashe et al., 2013).
To complement the OV model system, we recently reported the generation of a new virus-C. elegans model that uses the negative-sense, ssRNA vesicular stomatitis virus (VSV) (Gammon et al., 2017). Infection of wild-type (N2) worms with VSV is lethal although mutants defective in antiviral responses (e.g., drh-1 mutants) succumb to infection more rapidly (Gammon et al., 2017). Therefore, one can use lifespan assays as a convenient phenotypic readout when comparing different worm backgrounds for virus susceptibilities. Furthermore, the use of recombinant VSV strains encoding fluorescent reporters facilitates the scoring and tracking of infection in C. elegans tissues in real-time (Gammon et al., 2017). In addition, infection of worms with firefly luciferase-encoding VSV recombinants allows one to score virus replication using simple and quantitative luminescence assays (Gammon et al., 2017). Finally, the current study of VSV in a broad range of other model organisms (e.g., Drosophila, mice, etc.) provides the opportunity to examine VSV interactions with multiple invertebrate and vertebrate hosts. Here we describe how to establish VSV infection in C. elegans and use simple fluorescence and luminescence-based assays to track infection with the goal of uncovering nematode genetic backgrounds with differential susceptibilities to infection.
Materials and Reagents
- Personal protective equipment (gloves, lab coat, eye protection)
- Tissue culture dish, 150 x 25 mm (Corning, catalog number: 430599 )
- 50 ml conical tubes (Corning, catalog number: 431472 )
- Tissue culture dish, 6-well (Corning, catalog number: 3516 )
- 9” disposable borosilicate glass Pasteur pipets (Fisher Scientific, catalog number: 13-678-20C )
- FisherfinestTM Premium cover glasses (50 x 35 mm) (Fisher Scientific, catalog number: 12-548-5R )
- Glass needle, single capillary, 1.2 mm x 4 in. (102 mm) (World Precision Instruments, catalog number: 1B120F4 )
- Kimwipes (KCWW, Kimberly-Clark, catalog number: 34155 )
- Beckman Ultra-Clear ultracentrifuge tubes (Beckman Coulter, catalog number: 344058 )
- Eppendorf MicroloaderTM 20 µl pipette tips (Eppendorf, catalog number: 930001007 )
- 1.5 ml tube (VWR, catalog number: 20170-333 )
- 96-well plates (Corning, catalog number: 3915 )
- Modeling clay (Nasco, catalog number: 0300257M )
- C. elegans N2 strain (Caenorhabditis Genetics Center)
- Recombinant vesicular stomatitis virus encoding fluorescent marker gene [e.g., VSV-dsRED (Duntsch et al., 2004)] and/or firefly luciferase [e.g., VSV-LUC (Cureton et al., 2009)]
- Bacterial Escherichia coli strain OP50 (Caenorhabditis Genetics Center)
- Baby Hamster Kidney (BHK-21) cell line (ATCC, catalog number: CCL-10 )
- Vero cell line (ATCC, catalog number: CCL-81 ) or BSC-40 cell line (ATCC, catalog number: CRL-2761 )
- Methyl cellulose (Sigma-Aldrich, catalog number: 19-2930 )
- Crystal violet staining solution (Yamada and Takaoka, 2017)
- Crystal violet (Sigma-Aldrich, catalog number: C6158 )
- Agarose (Fisher Scientific, catalog number: BP160-500 )
- Microinjection oil (Series 700 Halocarbon oil) (Sigma-Aldrich, catalog number: H8898 )
- Reporter lysis buffer 5x (Promega, catalog number: E3971 )
- Luciferase Assay Reagent (Promega, catalog number: E1483 )
- 6.0% sodium hypochlorite solution (Fisher Scientific, catalog number: SS290 )
- Potassium hydroxide pellets (KOH) (Fisher Scientific, catalog number: P250 )
- Dulbecco’s modified Eagle’s medium (DMEM) (Sigma-Aldrich, catalog number: D6429 )
- Fetal bovine serum (FBS) (Atlanta Biologicals, catalog number: S12450 )
- Antibiotic-antimycotic solution, 100x (Sigma-Aldrich, catalog number: A5955 )
- L-Glutamine, 100x (Mediatech, catalog number: 25-005-CI )
- MEM nonessential amino acids (Mediatech, catalog number: 25-025-CI )
- NGM plates (He, 2011a)
- 5-Fluorodeoxyuridine (FUdR) (Sigma-Aldrich, catalog number: F0503 )
- 50% (w/v) polyethylene-glycol (PEG) 3000 (Rigaku Reagents, catalog number: 1008056 )
- M9 buffer (He, 2011a)
- Acetic acid (Fisher Scientific, catalog number: A38 )
- Methanol (PHARMCO-AAPER, catalog number: 339000000 )
- Formaldehyde (Fisher Scientific, catalog number: BP531-500 )
- Fixation solution (Yamada and Takaoka, 2017)
- Bleach solution (see Recipes)
- Complete DMEM (see Recipes)
- NGM + [50 µg/ml] FUdR Plates (see Recipes)
- M-PEG (see Recipes)
Equipment
- 37 °C cell culture incubator with 5% CO2 (Eppendorf, model: Galaxy® 170 S )
- Type 2 Biosafety cabinet (NuAire, model: NU-425-400 )
- Refrigerated benchtop centrifuge (e.g., Eppendorf, model: 5810 R ) with swinging bucket rotor capable of holding 50 ml conical tubes (e.g., Eppendorf, model: A-4-62 )
- Ultracentrifuge (e.g., Beckman Coulter, model: OptimaTM LE-80K ) with Beckman SW28 rotor (Beckman Coulter, model: SW 28 Ti )
- Refrigerated microcentrifuge (e.g., Southwest Science, model: SC1024-R )
- 50 °C water bath (e.g., VWR, model: 89501-468 )
- 80 °C oven (Gruenberg, model: CG45V240SS )
- Needle Puller (NARISHIGE, catalog number: PN-30 ) with Platinum board 3 mm filament (NARISHIGE, catalog number: PN-3H )
- Dissecting stereomicroscope (e.g., Nikon Instruments, model: SMZ745 )
- Refrigerated incubator capable of maintaining 15 °C (Sheldon Manufacturing, Shel Lab, model: SRI20 )
- Incubator capable of maintaining 25 °C (Sheldon Manufacturing, model: Model 2005 )
- Fluorescence stereo zoom microscope with dsRED and GFP filters (e.g., ZEISS, model: Axio Zoom.V16 )
- -80 °C freezer (VWR, model: VWR40086A )
- Bioruptor® II Type 12 (Cosmo Bio, model: Bioruptor2 Type 12 , catalog number: TOS-BR2012A)
- Envision 2102 Multilabel Reader (PerkinElmer, catalog number: 2105-0010 )
- Platinum Wire for worm pick, 30 gauge 0.254 mm diameter (Genesee Scientific, catalog number: 59-30P6 )
- Worm pick handle (Genesee Scientific, catalog number: 59-AWP )
- Autoclave (e.g., Getinge, model: 633LS )
- Air Table (e.g., Kinetic Systems, model: VIBRAPLANE )
- Compressed Nitrogen Tank (for connection to air table and microinjector unit) (e.g., Airgas, model: CGA-580 , catalog number: NI-NF300)
- Microinjector Unit (e.g., Eppendorf, model: FemtoJet® 5247 )
- Inverted microscope (e.g., Carl Zeiss, model: Axiovert 200 )
- Micromanipulator (e.g., Eppendorf, model: PatchMan 5173 )
- Micromanipulator controller (e.g., Eppendorf, model: 5171 )
Software
- Wallac EnVision Manager software for Envision 2102 Multilabel Reader (version 1.12)
- GraphPad Prism (v.6.0c, GraphPad Software Inc.)
Procedure
- Preparation of Vesicular Stomatitis Virus (VSV) stocks
Note: VSV is a Biosafety Level 2 (BSL-2) pathogen and thus should be handled using proper personal protective equipment (gloves, lab coat, eye protection). The virus can be inactivated and equipment decontaminated using either 70% ethanol or 10% Bleach solutions. Therefore, it is important to soak plastic ware, media, and needles exposed to virus in 10% Bleach for a minimum of 30 min prior to discarding in appropriate biohazard waste receptacles that are further decontaminated through autoclaving.- Seed ~5 x 106 BHK cells/dish into four 150 x 25 mm tissue culture dishes. Allow cells to attach overnight which should result in dishes that are 80-90% confluent by the next day (Figure 1).
Note: Confluency can affect VSV titers and the optimal confluency to initiate infection is when cells cover 80-90% of the plate surface. - The following day, dilute starter VSV stock into 20 ml of DMEM with the appropriate volume (dependent upon titer of starting stock) to achieve a multiplicity of infection (MOI) of 0.01 using 5 ml of this VSV-DMEM mixture per dish for infection. For example, assuming a cell doubling time of ~24 h (producing a total cell number of 1 x 107/dish), 1 x 105 PFU would be needed per 5 ml of DMEM for infection of each dish.
- Incubate cells at 37 °C with 5% CO2 for 2 h, rocking every 30 min to spread the inocula evenly across the dish.
- 2 h post-infection (hpi) replace inocula with 25 ml of complete DMEM (see Recipes).
- After 24-36 hpi, when significant cytopathic effect has been observed such that ~50% of the cell monolayer has lifted off the plate and cells have become rounded in appearance (Figure 1), collect supernatants from infected cultures and place in 50 ml conical tubes.

Figure 1. BHK cell culture before (A and C) and after ~30 h of VSV infection (B and D) - Spin harvested supernatants in a benchtop centrifuge with a swinging bucket rotor at 1,000 x g for 10 min at 4 °C to pellet cells.
- Transfer equal volumes of cleared supernatants (~25 ml/each) to Beckman Ultra-Clear ultracentrifuge tubes and place tubes in an SW28 swinging bucket rotor.
- Pellet virus particles by ultracentrifugation at 25,000 rpm (~50,000 x g) for 2 h at 4 °C. Pellets should be observable at the bottom of each tube after ultracentrifugation.
- Aspirate supernatants and resuspend virus particles in 0.5 ml or less DMEM.
- Aliquot 10 µl of concentrated VSV stock into cryovials and freeze at -80 °C.
Note: It is important to freeze multiple, small aliquots of VSV as virus titers rapidly decrease after multiple freeze-thaw cycles and thus each tube should be thawed for a single use. - To determine the number of plaque-forming units (PFU)/ml in the resulting VSV stock, titer the concentrated stock using 10-fold serial dilutions and plaque assays in 6-well dishes of Vero (or BSC-40) cells (Yamada and Takaoka, 2017). Briefly, ~5 x 106 Vero cells are seeded in 6-well dishes and allowed to come to confluence and then are infected for 2 h with serial dilutions of the VSV stock in (usually ranging from 10-5 to 10-9) and overlaid with 2 ml of complete DMEM containing 2.4% methyl cellulose for 24 h. The cells are then formaldehyde-fixed using fixation solution (Yamada and Takaoka, 2017), the overlaid media is removed and plaques are visualized with crystal violet staining. For more details and recipes for plaque assays, see Yamada and Takaoka, 2017.
Note: VSV titers of ~1 x 1010 PFU/ml are typically obtained with this method.
- Seed ~5 x 106 BHK cells/dish into four 150 x 25 mm tissue culture dishes. Allow cells to attach overnight which should result in dishes that are 80-90% confluent by the next day (Figure 1).
- Preparation of agarose pads
- Boil a solution of 2% agarose in water and place in a 50 °C water bath.
- Use a Pasteur pipette to transfer a drop of hot agarose onto the middle of a large coverslip, then immediately drop a second coverslip at a 90° angle to the first on top of the agarose drop.
- Once agarose has solidified, slide coverslips apart. Place the coverslip-pad in an 80 °C oven for one hour to dry the pad.
Note: See Pastuhov et al. (2017) for a demonstration of how to make an agarose pad.
- Boil a solution of 2% agarose in water and place in a 50 °C water bath.
- Preparing the needles
Note: The procedure below describes our methods for preparing needles for microinjection and the injection procedure itself. For additional details on microinjection see Evans, 2006.- Pull needles using glass capillary tubes in PN-30 needle puller.
Note: Determining the proper settings to pull the needles requires trial and error, as variations in capillary tube diameter and size and orientation of filament changes the quality of needles pulled. Good needles should easily pierce worms without killing them. - Slide the capillary tube through the filament ring using the grooves as a guide, being careful not to disturb the filament itself.
- Position the tube so the filament is halfway down then tighten the knobs on either side of the filament to hold the capillary tube in place (Figure 2A).

Figure 2. Preparation of needles for microinjection. A. Positioning of capillary tube in PN-30 needle puller; B. Resulting needle after needle pull; C. Setup for breaking pulled needle on microinjection scope; D. Positioning of needle next to cover glass edge prior to breaking; E. Bending of needle against cover glass edge to create opening; F. Confirmation of needle end opening by air bubble creation.
Note: Images in D-F are field of views at 40x magnification. - Close the cover lid and press the Start button. The pulling of the capillary tube should result in a needle with a pointed end (Figure 2B).
- The needles have closed tips and thus require breaking before use. Load a needle into the microinjection apparatus as if to inject worms (Figure 2C).
- Carefully wrap a cover glass in cloth or paper towel and gently bend the glass until it breaks. Ideally, break a 50 mm cover glass into 4-6 fragments.
- Place a fragment of a broken coverslip on top of a second coverslip with a drop of microinjection oil to keep the broken piece in place (Figure 2C).
- Place the coverslip on the stage and bring the edge of the glass fragment into focus using the 5x objective.
- Bring the needle into focus next to the glass fragment edge and switch to the 40x objective (Figure 2D).
- Refocus if necessary and gently push the needle against the side of the glass fragment. Once the needle tip bends slightly (Figure 2E) and vibrates against the glass, pull it away and inject a small amount of air into the oil. An air bubble confirms the tip has been broken open (Figure 2F).
- Pull needles using glass capillary tubes in PN-30 needle puller.
- Preparation of test animals
- Synchronize worms (N2 and test animals) in the L1 stage using bleach solution (see Recipes) as described (He, 2011b).
- Distribute roughly 50 to 100 L1 worms/genotype to test onto 35 mm NGM plates containing OP50 bacteria.
- Grow worms to young adult stage.
Notes:- Determine the optimal growth condition, including temperature, to assure that worms of different genetic backgrounds reach the young adult stage on the day of injection. We suggest seeding multiple plates with worms and keeping plates at multiple temperatures [e.g., 15 °C, room temperature (RT), 25 °C] as mutant animals may develop at different rates. We recommend roughly 96 h at 15 °C, or roughly 48 h at RT to generate young adults.
- In addition to N2 animals, it is recommended to include a hypersusceptible mutant to VSV such as drh-1 mutants, which have a defective antiviral RNAi response (Gammon et al., 2017), to ensure the virus stock is infectious and to compare test animals to both extremes of infection.
- Determine the optimal growth condition, including temperature, to assure that worms of different genetic backgrounds reach the young adult stage on the day of injection. We suggest seeding multiple plates with worms and keeping plates at multiple temperatures [e.g., 15 °C, room temperature (RT), 25 °C] as mutant animals may develop at different rates. We recommend roughly 96 h at 15 °C, or roughly 48 h at RT to generate young adults.
- Synchronize worms (N2 and test animals) in the L1 stage using bleach solution (see Recipes) as described (He, 2011b).
- Preparation of needles with injection mixture
- Place a strip of modeling clay inside a 150 x 25 mm Petri dish in order to hold needles.
- Thaw 10 µl 1 x 1010 PFU/ml VSV-dsRED or VSV-LUC on ice, then mix with 90 µl DMEM. Based on an estimated injection volume of ~10 nl/worm (using femtojet injection pressure and timing settings of 30 psi and 2 sec, respectively), this injection mixture results in the delivery of ~104 PFU/animal.
Note: Lower doses (102-103 PFU) can also establish infection although infection rates are dose-dependent (Gammon et al., 2017). - Centrifuge injection mixture at 5,000 x g at 4 °C for 10 min to pellet any particulates that could clog the needle. Transfer 20 µl of supernatant to a new 1.5 ml tube to use as injection media.
Note: DMEM lacking VSV is used for mock-infection media, which serves as a negative control for infection. - Lightly soak a Kimwipe with water, fold it into a strip and tape it to the inside of the needle-holding Petri dish to maintain humidity.
- Use microloader tips to load 2 µl of injection media (or DMEM for mock-infections) into 4-6 needles. Place each loaded needle into the needle container. An example of the needle holder setup is shown in Figure 3.
Note: To remove air bubbles in needles, let them set vertically (with the needle point facing downward) for a few minutes to allow gravity to remove bubbles. If air bubble persists, gently tap the side of the needle to agitate air bubble into escaping.
Figure 3. Needle holder setup. Needles are placed firmly into modeling clay within a 150 x 25 mm Petri dish that also contains a dampened Kimwipe to maintain humidity.
- Place a strip of modeling clay inside a 150 x 25 mm Petri dish in order to hold needles.
- Microinjection of virus media
Note: Because microinjection is a procedure that takes time to master, we suggest practicing by injecting worms with virus-free DMEM and ensuring that the vast majority (> 80%) of injected worms survive the next day before proceeding with VSV injections.- Dip a pipette tip in microinjection oil and spread a very thin layer of oil across the agarose pad of a coverslip.
Note: If the oil layer is too thick, worms will retain freedom of movement and it will take extra time to carefully press down the worm so that it settles and sticks in place on the agarose injection pad. Once the worm is on the pad, it begins to desiccate and will not survive injection if left too long. - Load a prepared needle into the needle holder and mount it on the manipulator as in Figure 2C.
- On a dissecting stereoscope, transfer one to several worms from their NGM plate to the agarose pad using a worm pick.
Note: To do so easily, first touch the pick to the microinjection oil, then use the oil to help pick up the worms using the bottom of your pick. Minimize the transfer of bacteria from the plate to the pad. - Use the pick to gently orient the worms all into a row where all the worms are facing the same direction.
- Transfer the slide to the microinjection scope and focus the worms using the 5x objective. Raise the level of focus to slightly above the worms, then use the manipulator to position the needle until its tip is in focus, right above the worms.
- Switch to the 40x objective, re-focus on the worms and then lower the needle into the field of view. The needle tip should be in focus next to the worm, at the focal plane where the worm is the widest.
- The goal is to insert the needle just posterior to the terminal bulb of the pharynx in order to inject virus into somatic (not gonadal) tissues (Figure 4). The needle should be inserted at a slight angle (15° to 40°) to ease its entry.

Figure 4. Demonstration of microinjection site - Inject the worms using femtojet injection pressure and timing settings of 30 psi and 2 sec, respectively.
- After injection, quickly transfer the coverslip back to the stereoscope. Pipette 5 µl of M-PEG (see Recipes) directly onto the worms in the oil. The worms will free themselves from the agarose and swim in the M-PEG. Once all worms are free, pipette up the M-PEG containing the worms, and then pipette the worms onto NGM plates containing Fluorodeoxyuridine (FUdR) (see Recipes).
Notes:- FUdR interferes with DNA replication and prevents the development of eggs and early larvae and thus allows injected animals to be tracked over several days without interference from worm progeny.
- Each FUdR plate should have 30-40 injected worms.
- FUdR interferes with DNA replication and prevents the development of eggs and early larvae and thus allows injected animals to be tracked over several days without interference from worm progeny.
- Once all injections have been performed, place all FUdR plates in an incubator at 15-25 °C.
Note: Virus replication and lifespan of infected nematodes are significantly altered by incubation temperature with lower temperatures typically resulting in fewer worms infected and greater survival (Gammon et al., 2017). We typically incubate infected worms at 25 °C to achieve higher rates of virus replication (Gammon et al., 2017).
- Dip a pipette tip in microinjection oil and spread a very thin layer of oil across the agarose pad of a coverslip.
- Scoring infection using fluorescence assay (after injection with VSV-dsRED)
Note: Worms injected with virus-free DMEM serve as a negative control group for infection in this experiment. These animals can be used to ensure fluorescence observed in VSV-dsRED-injected animals is not an artifact.- Worms that are unresponsive to a light touch by a platinum worm pick are considered dead and are removed from the experiment. All animals that die within 18 hpi are considered to have not recovered from the microinjection procedure itself and are removed from the experiment. Animals that crawl off the plate and are lost during the experiment are also censored. Scoring of the remaining worms is started 24 hpi.
- Using a fluorescent stereomicroscope, score worms for the presence of dsRED signal every 24 h and record the number of animals that are still alive, removing dead animals.
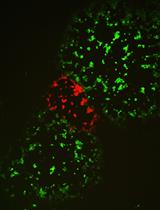
Note: N2 worms will typically present with dsRED signal in muscle tissue in the head, vulva, and tail (Figure 5). However, worms that are hypersusceptible to injection, such as drh-1 mutants, will display dsRED signals in multiple tissues throughout the body (Figure 5). Always check the GFP channel when scoring worms with apparent fluorescence signals to ensure the signal is not gut autofluorescence and that the signal is specific to the dsRED channel. Also, dsRED signals should be absent from the negative control group injected with just DMEM. Any animals with dsRED signal (in any tissue) that is clearly distinct from background autofluorescence observed in control animals are scored as positive for infection. Animals appearing to have different VSV susceptibilities can be further tested using luminescence assays (see below).
Figure 5. Differential interference contrast (DIC) and fluorescence micrographs of N2 and drh-1 animals infected with VSV-dsRED 72 hpi. Green fluorescence indicates autofluorescence signal in the intestine. See also Gammon et al., 2017 for more examples.
- Worms that are unresponsive to a light touch by a platinum worm pick are considered dead and are removed from the experiment. All animals that die within 18 hpi are considered to have not recovered from the microinjection procedure itself and are removed from the experiment. Animals that crawl off the plate and are lost during the experiment are also censored. Scoring of the remaining worms is started 24 hpi.
- Scoring infection using luminescence assay (after injection with VSV-LUC)
Note: Worms injected with virus-free DMEM serve as a negative control group for infection in this experiment.- At specific intervals post-infection (typically 24-72 hpi), worms should be harvested from each treatment to score for luciferase activity, which is used as a readout for VSV-LUC replication (Gammon et al., 2014 and 2017).
- At each time point chosen, pick 10 worms for each strain/treatment in 100 µl of reporter lysis buffer (in triplicate).
- Snap freeze in a -80 °C freezer or using liquid nitrogen, thaw at room temperature, sonicate using a Bioruptor® II sonicator (30 sec on high setting), snap freeze again, thaw at room temperature again.
- Spot 20 µl of lysates into a 96-well dish and mix each well with 100 µl Luciferase Assay Reagent.
- Measure arbitrary light units (LU) using Envision 2102 Multilabel Reader with Wallac EnVision Manager software.
Note: Lysates from worms injected with virus-free DMEM (control group) can be used to determine the background LU signals in order to subtract the average of these values from LU signals obtained from the lysates of VSV-LUC-challenged animals.
- At specific intervals post-infection (typically 24-72 hpi), worms should be harvested from each treatment to score for luciferase activity, which is used as a readout for VSV-LUC replication (Gammon et al., 2014 and 2017).
Data analysis
- Fluorescence assays
- After at least three independent experiments have been performed with (~30-40 injected worms/experiment), use GraphPad Prism software to plot the mean percentage of animals that score as infected (dsRED positive) over time (for example, see Figure 6A). Unpaired Student’s t-tests can be used to compare the maximum infection rates between two strains (e.g., N2 vs. drh-1) as described (Gammon et al., 2017).
Note: Depending upon the genotype of the worm, not all injections lead to an observable (e.g., dsRED-positive) infection but differences in the frequency of dsRED-positive animals after injection may be meaningful because it can suggest different susceptibilities to infection (e.g., compare N2 and drh-1 infection rates in Figure 6A). Also, by plotting the time at which maximum infection rates are reached in each genotype (e.g., in Figure 6A: N2 = 72 hpi, drh-1 = 48 hpi) one can also observe that more susceptible strains reach this maximum earlier than N2 strains. These data can be helpful in identifying genetic backgrounds with altered VSV susceptibilities. - Use GraphPad Prism software to plot the percentage of animals surviving over time (for example, see Figure 6B) to estimate the lifespan of the animals (from time of microinjection until death).

Figure 6. Example data showing percentage of nematodes (n = 30/group) scoring as infected (dsRED-positive) over time (A) and their survival (B) after infection with 103 PFU of VSV-dsRED - If desired, use non-linear regression analyses of survival curves to calculate the time at which 50% of the animals have died or ‘Lethal Time 50’ and their 95% confidence intervals as described (Gammon et al., 2017). Treatments that have non-overlapping 95% confidence intervals are considered to have significantly different lifespans. Alternatively, one can use GraphPad Prism software to construct Kaplan-Meier survival curves, and compare these with the logrank test (or the Wilcoxon-Gehan-Breslow test).
- After at least three independent experiments have been performed with (~30-40 injected worms/experiment), use GraphPad Prism software to plot the mean percentage of animals that score as infected (dsRED positive) over time (for example, see Figure 6A). Unpaired Student’s t-tests can be used to compare the maximum infection rates between two strains (e.g., N2 vs. drh-1) as described (Gammon et al., 2017).
- Luciferase assays
- After at least three independent experiments have been performed, use GraphPad Prism software to plot the mean LU recovered from each infected group at each time point (For example, see Figure 7).
- If desired, unpaired Student’s t-tests can be used to compare the mean LU obtained for two different strains for each time point as described (Gammon et al., 2017).

Figure 7. Example data showing light units (LU) obtained from lysates of nematodes infected with 104 PFU of VSV-LUC at the indicated times post-infection. These data demonstrate the elevated LU signals typically observed in LUC assays using lysates prepared from VSV-LUC-infected drh-1 animals in comparison to infected N2 worms. Data represent means of three independent experiments and error bars represent standard errors.
- After at least three independent experiments have been performed, use GraphPad Prism software to plot the mean LU recovered from each infected group at each time point (For example, see Figure 7).
Recipes
- Bleach solution
60 ml 6.0% sodium hypochlorite solution
30 ml 5 N KOH
410 ml ddH2O
Combine and store at 4 °C - Complete DMEM
500 ml DMEM
50 ml FBS
5 ml antibiotic-antimycotic
5 ml L-glutamine, 100x
5 ml MEM nonessential amino acids - NGM + [50 µg/ml] FUdR plates
Prepare NGM agar solution as described (He, 2011a) and add 50 µg/ml of FUdR prior to pouring. Store plates at 4 °C - M-PEG (100 ml)
0.1 ml 0.1% PEG 3000
100 ml M9 buffer
Combine and store at 4 °C
Acknowledgments
This protocol was adapted from Gammon et al., 2017. DG was supported by funding from the University of Texas Southwestern Medical Center’s Endowed Scholars Program. RL was supported by funding from the National Institutes of Health (grant GM84198). The authors thank Drs. Michael Whitt (The University of Tennessee Health Science Center) and Sean Whelan (Harvard Medical School) for the provision of VSV-dsRED and VSV-LUC. The authors declare no conflicts or competing interests.
References
- Ashe, A., Belicard, T., Le Pen, J., Sarkies, P., Frezal, L., Lehrbach, N. J., Felix, M. A. and Miska, E. A. (2013). A deletion polymorphism in the Caenorhabditis elegans RIG-I homolog disables viral RNA dicing and antiviral immunity. Elife 2: e00994.
- Cohen, L. B. and Troemel, E. R. (2015). Microbial pathogenesis and host defense in the nematode C. elegans. Curr Opin Microbiol 23: 94-101.
- Cureton, D. K., Massol, R. H., Saffarian, S., Kirchhausen, T. L. and Whelan, S. P. (2009). Vesicular stomatitis virus enters cells through vesicles incompletely coated with clathrin that depend upon actin for internalization. PLoS Pathog 5(4): e1000394.
- Duntsch, C. D., Zhou, Q., Jayakar, H. R., Weimar, J. D., Robertson, J. H., Pfeffer, L. M., Wang, L., Xiang, Z. and Whitt, M. A. (2004). Recombinant vesicular stomatitis virus vectors as oncolytic agents in the treatment of high-grade gliomas in an organotypic brain tissue slice-glioma coculture model. J Neurosurg 100(6): 1049-1059.
- Evans, T. C. (2006). Transformation and microinjection. In: WormBook (Ed.). The C. elegans Research Community. WormBook.
- Felix, M. A., Ashe, A., Piffaretti, J., Wu, G., Nuez, I., Belicard, T., Jiang, Y., Zhao, G., Franz, C. J., Goldstein, L. D., Sanroman, M., Miska, E. A. and Wang, D. (2011). Natural and experimental infection of Caenorhabditis nematodes by novel viruses related to nodaviruses. PLoS Biol 9(1): e1000586.
- Gammon, D. B. (2017). Caenorhabditis elegans as an emerging model for virus-host interactions. J Virol.
- Gammon, D. B., Duraffour, S., Rozelle, D. K., Hehnly, H., Sharma, R., Sparks, M. E., West, C. C., Chen, Y., Moresco, J. J., Andrei, G., Connor, J. H., Conte, D., Jr., Gundersen-Rindal, D. E., Marshall, W. L., Yates, J. R., Silverman, N. and Mello, C. C. (2014). A single vertebrate DNA virus protein disarms invertebrate immunity to RNA virus infection. Elife 3:e02910.
- Gammon, D. B., Ishidate, T., Li, L., Gu, W., Silverman, N. and Mello, C. C. (2017). The antiviral RNA interference response provides resistance to lethal arbovirus infection and vertical transmission in Caenorhabditis elegans. Curr Biol 27(6): 795-806.
- He, F. (2011a). Common worm media and buffers. Bio Protoc e55.
- He, F. (2011b). Synchronization of worm. Bio Protoc e56.
- Pastuhov, S. I., Shimizu, T. and Hisamoto, N. (2017). Heavy metal stress assay of Caenorhabditis elegans. Bio Protoc e2312.
- Yamada, T. and Takaoka, A. (2017). FICZ exposure and viral infection in mice. Bio Protoc e2096.
- Zhang, R. and Hou, A. (2013). Host-microbe interactions in Caenorhabditis elegans. ISRN Microbiol 2013: 356451.
Article Information
Copyright
© 2017 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Martin, A., Rex, E. A., Ishidate, T., Lin, R. and Gammon, D. B. (2017). Infection of Caenorhabditis elegans with Vesicular Stomatitis Virus via Microinjection. Bio-protocol 7(22): e2617. DOI: 10.21769/BioProtoc.2617.
Category
Microbiology > in vivo model > Viruses
Immunology > Animal model > Other
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.