- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Isolation, Culture and Differentiation of Adult Hippocampal Precursor Cells
(*contributed equally to this work) Published: Vol 7, Iss 21, Nov 5, 2017 DOI: 10.21769/BioProtoc.2603 Views: 20134
Reviewed by: Anonymous reviewer(s)
Original research article
The authors used this protocol in:

Mar 2017
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
There are two neurogenic niches in the adult mammalian brain: the subventricular zone of the lateral ventricle and the subgranular zone of the hippocampal dentate gyrus. Cells from these areas can be isolated and maintained in vitro, using two different culture systems to assess their potential regarding proliferation and differentiation in a reductionist model. While the neurosphere assay is primarily performed to directly study the proliferative and differentiation potential of cells in individual brains, the monolayer culture allows single cell analysis in a rather homogeneous cell population. Here, we describe the isolation, culturing methods and differentiation of neural precursor cells in both systems.
Keywords: NeuroscienceBackground
In the mammalian brain, adult neural stem cells reside in two main neurogenic niches, the subgranular zone (SGZ) of the hippocampal dentate gyrus (DG) and the lateral ventricle of the subventricular zone (SVZ), that allow the generation of new neurons in the adult brain. Neural precursor cells from the neurogenic niches can be isolated and cultured in vitro to model cellular processes, especially proliferation and differentiation. Two standard culture systems, the adherent monolayer culture (Palmer et al., 1995; Ray et al., 1995) and the neurosphere assay (Reynolds and Weiss, 1992 and 1996), both introduced in the 1990s, represent valuable tools to study neural progenitor cell biology in vitro.
Depending on the research question, each system has advantages and disadvantages that should be considered carefully before choosing one or the other culture method. In adherent monolayer cultures cells grow rather isolated and form more homogeneous cultures. Monolayers allow the direct investigation and monitoring of neural precursor cells at the single cell level. Characteristics like morphology, proliferation and differentiation under controlled conditions, can easily be analysed and visualised. However, compared to neurosphere cultures, cells cultured as monolayer represent a more reductionist model as the cells grow with fewer cell-to-cell contacts that are usually present in the niche.
Neurosphere cultures are free-floating aggregate cultures that are easy to obtain from adult tissue. Primary neurospheres are more heterogeneous and presumably represent a more niche-like environment. Neurospheres can be used to model the interaction of different cell types and allow relative comparisons of precursor cell number and potential, but does not allow absolute conclusions about stem cell numbers in vivo. Also, the sphere-forming capacity is not identical to ‘stemness’.
This protocol describes the detailed workflow of the generation and analysis of adult neural precursor cultures as neurospheres and monolayers from both neurogenic regions, the SVZ and the DG. The protocol represents an optimized version of our previously published protocols that have been successfully applied to many research projects within our group and by other groups (Babu et al., 2011; Walker and Kempermann, 2014; Ehret et al., 2016; Hörster et al., 2017).
Materials and Reagents
- Animals
Mice: C57BL/6J (8 weeks old)
Note: We recommend three to four mice for establishing a monolayer cell culture. For neurosphere assay experiments we recommend one mouse per 96-well plate. - General materials and reagents
- Centrifugation tubes 15 ml and 50 ml
- Reaction tubes 1.5 ml
- Parafilm
- 70% ethanol
- Double distilled water (ddH2O)
- 1x phosphate-buffered saline (PBS)
- DMEM/F-12 without glutamine (Thermo Fisher Scientific, GibcoTM, catalog number: 21331020 )
- 4% paraformaldehyde (PFA) in 0.1 M phosphate buffer pH 7.4
- PFA (Merck, catalog number: 1040051000 )
- Sodium dihydrogen phosphate (Merck, catalog number: 1063421000 )
- Disodium phosphate dihydrate (Acros Organics, catalog number: 343810025 )
- Sodium hydroxide (NaOH) (Carl Roth, catalog number: 6771 )
- PFA (Merck, catalog number: 1040051000 )
- Growth media (see Recipes)
- Neurobasal® medium (Thermo Fisher Scientific, GibcoTM, catalog number: 21103049 )
- B-27® supplement (50x) (Thermo Fisher Scientific, GibcoTM, catalog number: 17504044 )
- Pen/Strep 100,000 U/ml (Thermo Fisher Scientific, GibcoTM, catalog number: 15140122 )
- GlutaMAXTM supplement (100x stock) (Thermo Fisher Scientific, GibcoTM, catalog number: 35050061 )
- Neurobasal® medium (Thermo Fisher Scientific, GibcoTM, catalog number: 21103049 )
- Fire polished pipettes
- Glass Pasteur pipette (1 mm diameter)
- Glass Pasteur pipette (1 mm diameter)
- Coating
- Poly-D-Lysine hydrobromide (PDL) (Sigma-Aldrich, catalog number: P7280 )
- Laminin (Roche Diagnostics, catalog number: 11243217001 )
- Poly-D-Lysine hydrobromide (PDL) (Sigma-Aldrich, catalog number: P7280 )
- Brain dissection
Petri dishes (10 cm diameter) - SVZ tissue dissociation
- Petri dishes (6 cm diameter)
- Scalpel (#10) (Fisher Scientific, catalog number: 11995756)
Manufacturer: B. Braun Melsungen, catalog number: 5518059 . - Falcon® 40 µm cell strainer (Corning, Falcon®, catalog number: 352340 )
- 0.05% trypsin-EDTA (Thermo Fisher Scientific, GibcoTM, catalog number: 25300054 )
- Trypsin inhibitor containing DNAse I (see Recipes)
- Trypsin inhibitor (Sigma-Aldrich, catalog number: T6522 )
- DNase I (Roche Diagnostics, catalog number: 10104159001 )
- Trypsin inhibitor (Sigma-Aldrich, catalog number: T6522 )
- Petri dishes (6 cm diameter)
- DG tissue dissociation
- Petri dishes (6 cm diameter)
- Falcon® 40 µm cell strainer (Corning, Falcon®, catalog number: 352340 )
- Neural tissue dissociation kit (P) (Miltenyi Biotech, catalog number: 130-092-628 )
- Beta-mercaptoethanol (Sigma-Aldrich, catalog number: M7522 )
Note: This product has been discontinued. - Hank’s buffered salt solution (HBSS) (Thermo Fisher Scientific, GibcoTM, catalog number: 14175 )
- Petri dishes (6 cm diameter)
- Monolayer culture
- Tissue culture flasks (T25 and T75)
- 24-well tissue culture plates (growth-enhance treated, gamma-sterilized, free of pyrogens, free of RNA/DNA, DNase, RNase)
- Coverglass slips 12 mm (Fisher Scientific, catalog number: 12-545-82 )
Note: This product has been discontinued. - Heparin (MP Biomedicals, catalog number: 0210193125 )
- Human EGF (PeproTech, catalog number: AF-100-15 )
- Human FGF2 (PeproTech, catalog number: 100-18B )
- Accutase solution (Sigma-Aldrich, catalog number: A6964 )
- Dimethyl sulfoxide (DMSO) (Sigma-Aldrich, catalog number: D8418 )
- Normal donkey serum (Jackson ImmunoResearch Laboratories, catalog number: 017-000-121 )
- Trypan blue solution, 0.4% (Thermo Fisher Scientific, GibcoTM, catalog number: 15250061 )
- BrdU (Sigma-Aldrich, catalog number: B5002 )
- Freezing mix (see Recipes)
- Antibody solution (see Recipes)
- Tissue culture flasks (T25 and T75)
- Neurosphere assay
- Petri dishes (10 cm diameter)
- 96-well tissue culture plates (growth-enhance treated, gamma-sterilized, free of pyrogens, free of RNA/DNA, DNase, RNase)
- 24-well tissue culture plates (growth-enhance treated, gamma-sterilized, free of pyrogens, free of RNA/DNA, DNase, RNase)
- Coverglass slips 12 mm (Fisher Scientific, catalog number: 12-545-82 )
- Heparin (MP Biomedicals, catalog number: 0210193125 )
- Human EGF (Peprotech, catalog number: AF-100-15 )
- Human FGF2 (PeproTech, catalog number: 100-18B )
- Blocking solution (see Recipes)
- Petri dishes (10 cm diameter)
- Staining reagents
- Microscope slides SuperFrost® (VWR, Thermo Scientific, catalog number: 631-0706 )
- Triton® X-100 (Carl Roth, catalog number: 3051 )
- 1 N HCl (from 37% stock solution) (Sigma-Aldrich, catalog number: 435570 )
Note: This product has been discontinued. - 0.9% NaCl
- Normal donkey serum (Jackson ImmunoResearch Laboratories, catalog number: 017-000-121 )
- Hoechst 33342 (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 62249 )
- Primary antibodies (see Table 1)
- Aqua-Poly/Mount (Polysciences, catalog number: 18606 )
- Borate buffer (see Recipes)
- Boric acid (Carl Roth, catalog number: 6943.1 )
- Sodium hydroxide (NaOH) (Carl Roth, catalog number: 6771 )
Table 1. Primary antibodies for immunocytochemistryAntibody Host Clone Isotype Company Catalog number β-III-tubulin (β-tubulin) Mouse 5G8 IgG1 Promega G7121 5’-bromo-2’-deoxyuridine (BrdU) Rat BU1/75 (ICR1) IgG2a Bio-Rad Laboratories OBT0030 Glial fibrillary acidic protein (GFAP) Rabbit polyclonal - Agilent Technologies Z0334 Map2ab Mouse AP-20 IgG1 Sigma-Aldrich M1406 Nestin Mouse 25/NESTIN IgG1, κ BD 611658 Oligodendrocyte marker 4 (O4) Mouse O4 IgM R&D Systems MAB1326 Sox2 Rabbit polyclonal - Merck AB5603
- Boric acid (Carl Roth, catalog number: 6943.1 )
- Microscope slides SuperFrost® (VWR, Thermo Scientific, catalog number: 631-0706 )
Equipment
- Bunsen burner
- Autoclave
- Dissection tools
- Scissors
- Small spatula (Fine Science Tools, catalog number: 10093-13 )
- Curved forceps (Fine Science Tools, model: Dumont #7, catalog number: 11271-30 )
- Angled forceps (Fine Science Tools, model: Dumont #5-45, catalog number: 11253-25 )
- 27 G ¾ needle (B. Braun Melsungen, catalog number: 4657705-02 )
- Scissors
- Vacuum pump
- Stereo microscope (Olympus, model: SZ61 )
- Inverted microscope (Olympus, model: CKX42 )
- Centrifuge with swing bucket rotor for 15 ml and 50 ml centrifuge tubes (Eppendorf, model: 5430 R )
- Incubator at 37 °C with 5% CO2
- Sterile laminar flow hood
- Hemocytometer (Neugebauer improved)
- Fluorescence microscope (ZEISS, model: Axio Imager.M2 )
- Freezing containers, Mr. FrostyTM (Thermo Fisher Scientific, Thermo ScientificTM, model: Mr. FrostyTM, catalog number: 5100-0001 )
- 37 °C water bath
- -80 °C freezer
- Pipettes
- Multistepper pipette (and tips)
Procedure
- General preparations
Coating of cell culture vessels
Monolayer cultures as well as the differentiation of neurospheres require poly-D-lysine (PDL)/Laminin coated surfaces for attachment.- Add the appropriate amount of PDL (5 µg/ml in ddH2O) and assure that the surface or the coverslips are fully covered (for volumes see Table 2).
- Incubate for at least 4 h or overnight at room temperature.
- Remove the solution and wash the vessels three times with ddH2O.
- Let them dry properly until no residual water remains.
- Add Laminin (5 µg/ml in cold DMEM/F-12) and incubate for at least 4 h or overnight at 37 °C.
- Either use them directly or store them at -20 °C until required.
Fire polished pipettes
During the isolation process, fire-polished pipettes with either small or medium size bores are required.- Rotate the glass Pasteur pipettes for about 3 sec over a hot blue flame of a Bunsen burner until the edges become rounded. Small Pasteur pipettes show an internal diameter of 0.3-0.4 mm while the medium size bore of the pipettes measures approximately 0.6-0.8 mm.
- Pipettes should be autoclaved prior to use.
- Add the appropriate amount of PDL (5 µg/ml in ddH2O) and assure that the surface or the coverslips are fully covered (for volumes see Table 2).
- Brain dissection
Note: Monolayer cultures require coated 24-well plates, which should be prepared at least two days prior to isolation.
The protocol for neural precursor cell isolation is based on the protocols of Babu et al. (2011), Walker and Kempermann (2014) (which also includes a video of the dissection procedure) and Ehret et al. (2016). Neurosphere assays can be performed using one mouse per neurosphere experiment (per 96-well plate). Monolayer cultures can also be generated using a single mouse, however we recommend pooling 3-4 brains.- Anesthetize 6-8 weeks old mice according to the appropriate institutional guidelines. Perform cervical dislocation.
- Spray the head with 70% ethanol to sterilize the area and to reduce the amount of fur adhering to the dissection tools and tissue.
- Decapitate the animal at the base of the brain stem using sharp scissors.
- Cut the skin sagittally along the midline approximately until a point between the eyes. Expose the skull.
- Place one blade of a small pair of scissors into each eye cavity to perform a coronal cut of the skull between the eyes. Afterwards, make two lateral cuts at the base of the skull, followed by a longitudinal cut along the sagittal suture.
- Expose the brain by peeling back the skull with a pair of forceps.
- Remove the brain from the skull by using a small spatula and place it into cold PBS.
- Transfer the brain into a 10 cm plastic Petri dish containing PBS (see Figure 1B).
- Place the Petri dish with the brains under a stereo microscope at low magnification and position the brain on its ventral surface. Remove the olfactory bulbs using fine angled forceps while holding the brain in position by the cerebellum.
SVZ dissection- Rotate the brain onto the dorsal aspect and make a coronal cut through the brain at the area of the optic chiasm using a scalpel (see Figure 1C).
- For microdissection of the SVZ (for more details see Azari et al. (2010) and Walker and Kempermann, 2014), have the rostral portion of the brain exposing the lateral ventricles of the cut coronal surface. Increase the magnification, remove and discard the septum using fine curved forceps.
- Dissect the SVZ by placing the tip of one blade of a pair of fine curved forceps in the lateral corner of the lateral ventricle immediately under the corpus callosum and the other roughly 1 mm into the tissue immediately adjacent to the ventricle. Press down the forceps towards the base of the dish and the ventral aspect of the ventricle to remove a small triangular piece of tissue (see Figure 1E). Place the dissected tissue into a small Petri dish on ice.
DG dissection- For microdissection of the DG (for more details see Hagihara et al. (2009) and Walker and Kempermann, 2014), place the caudal portion of the brain into the Petri dish and use a scalpel to cut along the longitudinal fissure (see Figure 1D).
- Carefully remove the cerebellum and the diencephalon using fine angled forceps.
- Refocus the microscope to visualize the borders around the DG. To remove the DG, use a 27 G ¾ needle and carefully slide along the border between the DG and Ammons’s horn.
- Free the DG from the surrounding tissue using fine angled forceps (see Figure 1F). Place the dissected tissue into a small Petri dish in 10 µl PBS on ice.
- Anesthetize 6-8 weeks old mice according to the appropriate institutional guidelines. Perform cervical dislocation.
- SVZ tissue dissociation
- Preheat 1 ml of 0.05% trypsin-EDTA in a 15 ml centrifuge tube in a water bath at 37 °C for 10-15 min.
- Mince the dissected SVZ tissue with a scalpel in a 6 cm Petri dish for about 1 min (see Figure 1G).
- Transfer tissue pieces into the warm trypsin-EDTA and incubate for 7 min at 37 °C. Gently mix by inverting the tube occasionally.
- Stop the enzyme reaction by adding 1 ml of trypsin inhibitor containing DNaseI (see Recipes) and mix gently by flicking the tube.
- Centrifuge for 5 min at 300 x g.
- Gently resuspend the pellet in 1 ml of growth medium (see Recipes) by carefully pipetting up and down 7 to 10 times using a P1000 pipette (see Figure 1H).
Note: To increased cell death, it is important to triturate the tissue very gently. - Add up to 5 ml of growth medium and pass the cell suspension through a 40 µm cell strainer into a 50 ml centrifuge tube.
- Centrifuge for 5 min at 300 x g.
- Resuspend the cell suspension in 500 µl of growth medium.
- Preheat 1 ml of 0.05% trypsin-EDTA in a 15 ml centrifuge tube in a water bath at 37 °C for 10-15 min.
- DG tissue dissociation
Note: The average yield of cells that can be obtained from one animal varies depending on the age of the animal as well as on the strain. However, to provide a rough estimate, we usually gain about 100,000-200,000 cells/ml from one 8-week old C57BL/6J animal.
To obtain a single-cell suspension from the isolated DG tissue, the Neural Tissue Dissociation Kit (P) containing papain is applied as follows:- To prepare Enzyme Mix 1 add 1,900 µl Buffer X and 50 µl Enzyme P (both supplied in the kit) to a 15 ml centrifuge tube. To increase the stability of the enzymes and viability of the cells add beta-mercaptoethanol to Buffer X to a final concentration of 0.067 mM.
- Preheat Enzyme Mix 1 in a water bath at 37 °C for 10-15 min.
- Mince the dissected DG tissue with a scalpel in a 6 cm Petri dish for about 1 min (see Figure 1G).
- Transfer the tissue pieces into 1,950 µl of preheated Enzyme Mix 1.
- Incubate for 15 min at 37 °C and gently mix by inverting the tube every 3-5 min.
- Prepare Enzyme Mix 2 by adding 20 µl of Buffer Y to 10 µl of Enzyme A (both supplied in the kit).
- Add Enzyme Mix 2 to the tissue-enzyme mix.
- Dissociate the tissue mechanically by gently pipetting up and down 10 x with a fire-polished Pasteur pipette with the medium bore.
Note: To avoid increased cell death, it is important to triturate the tissue very gently. - Incubate for 10 min at 37 °C and gently mix by inverting the tube every 3-5 min.
- Further dissociate the tissue mechanically using a small bore, fire-polished Pasteur pipette by gently pipetting up and down 10 x (see Figure 1I).
- To wash the cell suspension, add Hank’s buffered salt solution up to 10 ml.
- Centrifuge at 300 x g for 5 min.
- Resuspend the pellet in 500 µl of growth medium and apply to 40 µm cell strainer placed in a 50 ml centrifuge tube.
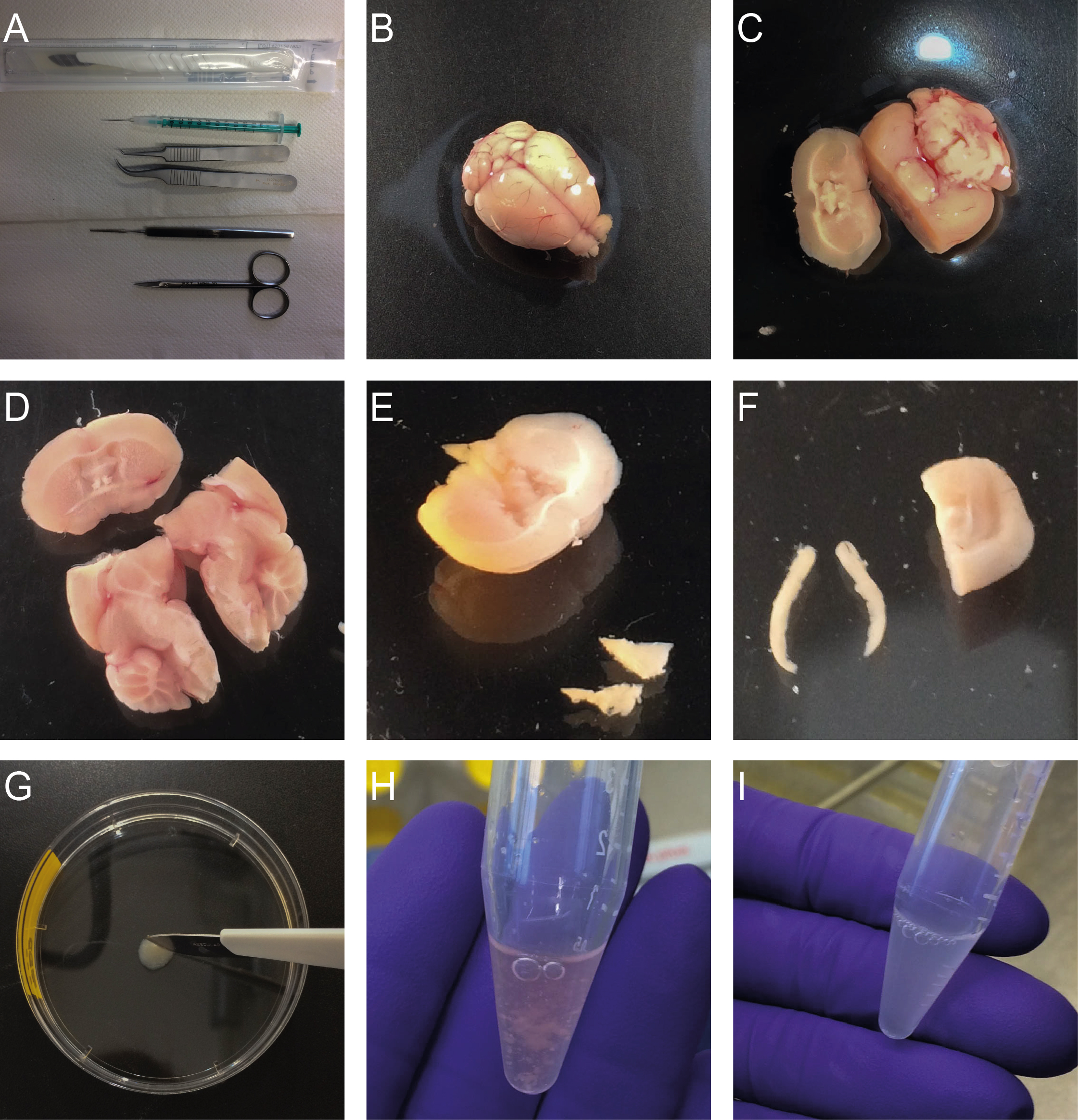
Figure 1. Isolation of neural precursor cells from the SVZ and the DG. A. Required tools; B. Isolated brain; C. Coronal cut through the brain at the area of the optic chiasm using a scalpel; D. Cut along the longitudinal fissure; E. Dissected SVZ tissue; F. Dissected DG; G. Mincing of the dissected tissue using a scalpel; H. Resuspended SVZ tissue; I. DG tissue after enzymatic incubation.
- To prepare Enzyme Mix 1 add 1,900 µl Buffer X and 50 µl Enzyme P (both supplied in the kit) to a 15 ml centrifuge tube. To increase the stability of the enzymes and viability of the cells add beta-mercaptoethanol to Buffer X to a final concentration of 0.067 mM.
- Monolayer culture
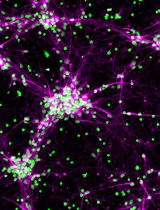
Monolayer cultures are established from primary cells, which are then cultured as adherent cells over several passages under proliferation conditions (Figure 2A). This leads to a rather homogeneous culture that can be used to perform single cell analyses.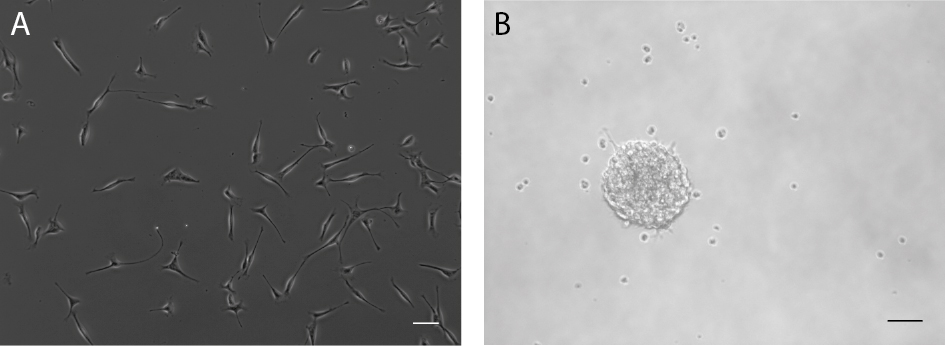
Figure 2. Neural precursor cells from the adult mouse brain can be cultured as adherent monolayer cultures (A) or as neurospheres (B). Scale bars = 50 µm.
Culturing- Add 20 ng/ml FGF2, 20 ng/ml EGF and 2 µg/ml Heparin to the single cell suspension in growth medium obtained from the tissue dissociation.
- Seed the cell suspension into a coated well of a 24-well plate.
- 24 h after seeding, remove the growth medium and exchange it with fresh growth medium with growth factors (20 ng/ml FGF2, 20 ng/ml EGF and 2 µg/ml Heparin).
Note: This step can be skipped if the cells don’t seem to be sufficiently attached. - 24 h later, wash the cells with prewarmed PBS and add fresh growth medium with growth factors (20 ng/ml FGF2, 20 ng/ml EGF and 2 µg/ml Heparin).
- Every other day, exchange 50% of the old growth medium with fresh growth medium containing 100% of the growth factors to counteract the growth factor consumption in the residual medium (e.g., for a T25 flask containing 5 ml medium, remove 2.5 ml and add 2.5 ml fresh medium containing 40 ng/ml EGF and FGF2 and 4 µg/ml Heparin).
- Once cells reach 70-80% confluency, remove the medium and wash once with PBS.
Note: This can take up to two weeks. - Add 150 µl of Accutase and incubate at 37 °C for 3 min.
- Tap the plate strongly onto its surface as well as from the side to agitate the attached cells.
- Check under the microscope if the cells are detached. If not, prolong incubation time (max 10 min).
- Add 2 ml of growth medium to the cell suspension and spin at 300 x g for 3 min to pellet the cells.
- Remove the supernatant and resuspend the cells in 1 ml of growth medium.
- Take out 10 µl of cell suspension and mix it with 10 µl of trypan blue.
- Count the cells with a hemocytometer.
- Seed the cells in a density of min. 1 x 104 c/cm2 in growth medium with growth factors (20 ng/ml FGF2 and 20 ng/ml EGF; no more Heparin) into a new vessel.
Further passaging (passage only once cells have reached 70-80% confluency)- Remove the medium and wash once with PBS.
- Add Accutase and incubate at 37 °C for 3 min.
- Tap the plate strongly onto its surface as well as from the side to agitate the attached cells.
- Check under the microscope if the cells are detached. If not, prolong incubation time (max 10 min).
- Collect the cells in 4.5 ml of growth medium (volume for a T25 flask) and spin at 300 x g for 3 min to pellet the cells.
- Add 10 µl of trypan blue to 10 µl of cell suspension and count cells with a hemocytometer.
- Seed the cells in a density of min. 1 x 104 c/cm2 (live cells) in growth medium containing growth factors (20 ng/ml FGF2 and 20 ng/ml EGF) into a new vessel.
Freezing- Prepare freezing mix (see Recipes) and store at 4 °C.
- Remove growth medium from flask/plate and wash with 5 ml of PBS.
- Add Accutase to the flask/plate and incubate for 3 min at 37 °C, check if all the cells are detached. If not, prolong incubation time (max 10 min).
- Collect cells in 4.5 ml of growth medium (volume for a T25 flask) and spin at 300 x g for 3 min to pellet the cells.
- Remove the supernatant.
- Resuspend cells in 1 ml of growth medium.
- Add 10 µl of trypan blue to 10 µl of cell suspension.
- Count the cells and adjust the cell density to 2 x 106 c/ml (live cells) with growth medium.
- Add 0.5 ml of freezing mix to each freezing vial.
- Add 0.5 ml of cell suspension to each freezing vial, mix and put into a freezing container.
- Place the freezing container directly in the -80 °C freezer.
- After 12 h, vials can be transferred into liquid nitrogen.
Cell seeding and analysis of cell characteristics under proliferation conditions (Figure 3)
Note: To analyse cell characteristics under proliferation conditions, cells can either be fixed directly 48 h after seeding or labeled with BrdU.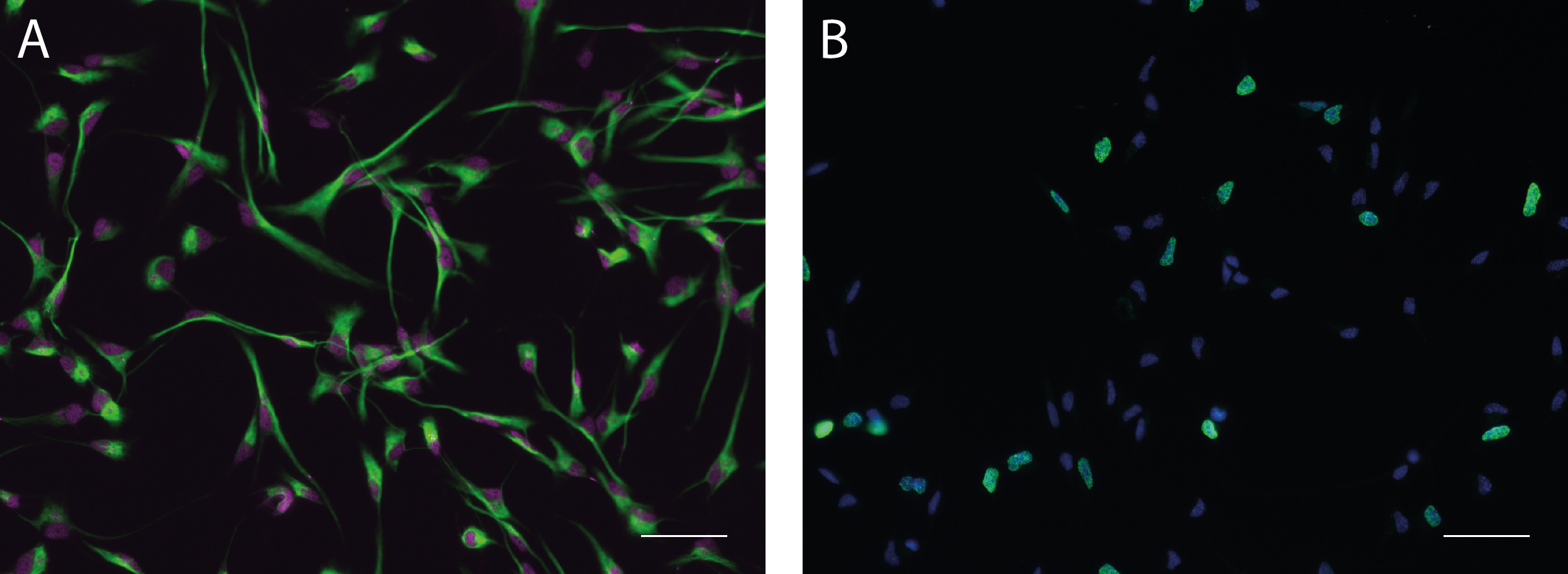
Figure 3. Monolayer cultures under proliferation conditions. A. Monolayer cultures under proliferation conditions express cellular markers for progenitor cells of the nervous system such as Nestin (green) and Sox2 (magenta). B. Progenitor cells can be labeled to assess their proliferation capacity by examining the fraction of cells undergoing S-phase (BrdU label, green) relative to the overall number of cells (Hoechst 33342, blue). Scale bars = 50 µm.- Thaw PDL/Laminin-coated plates (24-well plates with coverslips) for about 15 min at 37 °C.
- Remove the Laminin from the 24-well plates.
- Plate 20,000 cells/well onto coated coverslips in 24-well plate wells under proliferation conditions (add 20 ng/ml EGF and 20 ng/ml FGF2 to the medium)
- Incubate for 48 h.
- Add BrdU (final concentration 10 µM) to each well and incubate for 2 h at 37 °C.
Note: Skip this step if no BrdU labeling is required and continue with step 6. - Remove the medium, add 4% PFA in 0.1 M phosphate buffer to each well and incubate for 20 min at room temperature.
- Wash twice with PBS and add 1 ml of fresh PBS into the wells.
- Store at 4 °C until staining is performed. To avoid evaporation, seal the plate with Parafilm.
Cell seeding and differentiation (Figure 4A)- Thaw PDL/Laminin-coated plates (24-well plates with coverslips) for about 15 min at 37 °C.
- Remove the Laminin from the 24-well plates and wash the plate with PBS.
- Plate 20,000 cells/well onto coated coverslips in 24-well plate wells under proliferation conditions (add 20 ng/ml EGF and 20 ng/ml FGF2 to the medium)
- Incubate for 48 h.
- Remove growth medium and add fresh growth medium containing 5 ng/ml FGF2.
- After another 48 h, remove growth medium and add fresh growth medium without growth factors.
- Let the cells differentiate for 3-5 days.
Fixation- Remove the growth medium.
- Add 300 µl of 4% PFA in 0.1 M phosphate buffer to each well and incubate for 20 min at room temperature.
- Wash twice with PBS and add 1 ml of fresh PBS into the wells.
- Store at 4 °C until staining is performed. To avoid evaporation, seal the plate with Parafilm.
Staining
Note: Steps 1-4 are required for BrdU stainings only, for other stainings start directly with step 5.- Wash 2 x with 0.9% NaCl.
- Incubate for 30 min with 1 N HCl at 37 °C.
- Wash 1 x with Borate buffer (see Recipes).
- Rinse 3 x 10 min with PBS.
- Permeabilize with 0.1% Triton X-100 in PBS for 10 min.
- Block with blocking solution at room temperature for 1 h.
- Incubate with primary antibodies (see Table 1) in antibody solution for 2 h at room temperature otherwise overnight at 4 °C (300 µl/well).
- Wash 3 x with PBS.
- Incubate with appropriate secondary antibodies diluted in antibody solution (see Recipes) for 1 h at room temperature (300 µl/well) in the dark.
- Wash 10 min with PBS.
- Incubate with Hoechst 33342 (1:4,000 in PBS) for 10 min at room temperature in the dark.
- Wash 1 x with PBS.
- Dip-wash in ddH2O.
- Dry coverslip by gently tapping on tissue paper and mount with fluorescence mounting medium.
- Dry overnight in darkness.
- Store at 4 °C.
- Image using a fluorescence microscope.
- Add 20 ng/ml FGF2, 20 ng/ml EGF and 2 µg/ml Heparin to the single cell suspension in growth medium obtained from the tissue dissociation.
- Neurosphere assay
Neurosphere cultures are easy to generate and allow straightforward data analysis with regards to precursor cell numbers and potential in brain tissue derived from individual mice (Figure 2B).
Culture
Note: Neurospheres are cultured in growth medium that additionally contains 2 µg/ml Heparin.- Dilute the single cell suspension obtained from one mouse in 20 ml of neurosphere growth medium containing 20 ng/ml FGF2, 20 ng/ml EGF and 2 µg/ml Heparin.
- Plate 200 µl per well across a 96-well plate using a 10 ml multistepper pipette.
- Incubate SVZ-derived neurosphere cultures for 7 days and DG-derived neurosphere cultures for 12 days at 37 °C.
Note: Extended incubation times can lead to overgrowth and may result in spontaneous attachment, differentiation or cell death in the core of the neurospheres. - Count and size neurospheres using an inverted light microscope.
Differentiation (Figures 4B and 4C)
Note: For one experiment, we recommend seeding at least 4 coverslips with randomly sized neurospheres per 96-well plate.- Thaw PDL/Laminin-coated plates (24-well plates with coverslips) for about 15 min at 37 °C.
- Remove the Laminin from the 24-well plates and wash the plate with PBS.
- Add 1 ml of growth medium to each well.
- Collect the medium containing the neurospheres from all wells of the 96-well plate into a 10 cm Petri dish.
- Using a stereo microscope collect the neurospheres from the Petri dish with a P100 pipette (final volume of 75 µl) and transfer them onto the coverslips in the 24-well plates. Per coverslip we recommend to seed about 15 neurospheres.
- Differentiate the neurospheres for 7 days in the incubator at 37 °C. The cells will spread out over the coverslip and become adherent.
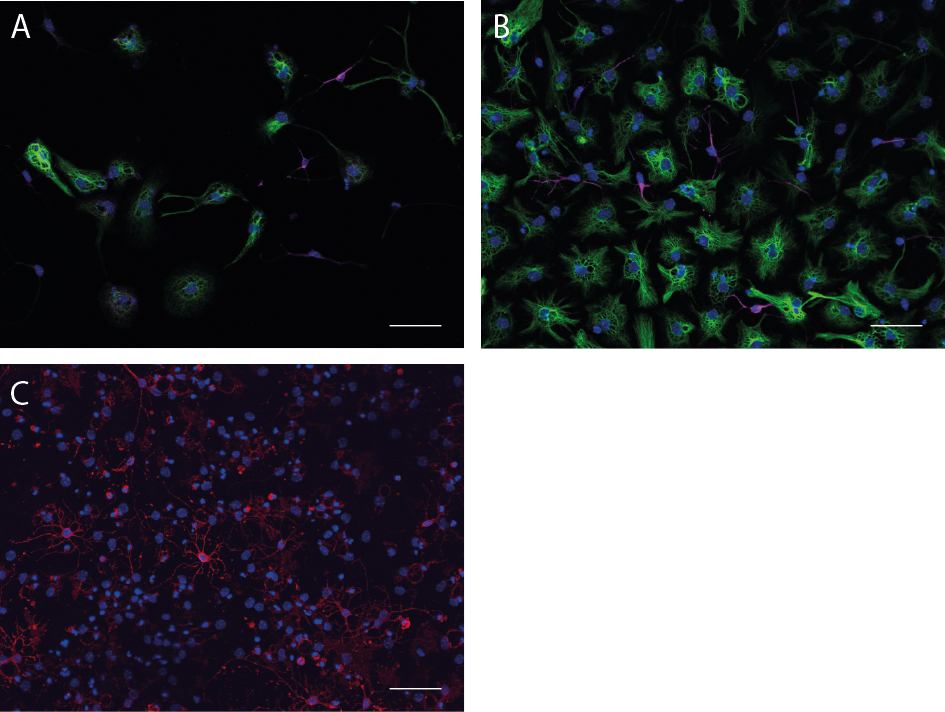
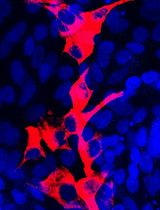
Figure 4. Monolayer cultures as well as neurospheres can be differentiated. A. Differentiated cells of a monolayer culture with GFAP-positive astrocytes (green) and Map2ab-positive neurons (magenta). B. Differentiated neurosphere culture with β-III-tubulin-positive neurons (magenta) and GFAP-positive astrocytes (green). C. Differentiated neurosphere culture with O4-positive Oligodendrocytes (red). Scale bars = 50 µm.
- Remove the medium and wash 2 x with PBS to remove dead cells and debris.
- Add 300 µl of 4% PFA in 0.1 M phosphate buffer to each well and incubate for 20 min at room temperature.
- Remove PFA and wash twice with PBS.
- Store in 1 ml PBS at 4 °C. To avoid evaporation, seal the plate with Parafilm.
Staining
Note: For staining with the recommended O4 antibody, do not add Triton to the blocking and antibody solutions and use an appropriate IgM secondary antibody.- Wash the coverslips containing the differentiated neurospheres 2 x with PBS.
- Incubate the coverslips in blocking solution (see Recipes) containing 0.2% Triton X-100 for 30 min at room temperature.
- Incubate with primary antibodies against the neuronal and astrocytic markers or the oligodendrocyte marker O4 (see Table 1) in antibody solution containing 0.2% Triton X-100 for 1 h at room temperature (300 µl/well).
- Wash 4 x with PBS.
- Incubate for 30 min with appropriate secondary antibodies in antibody solution containing 0.2% Triton X-100 at room temperature in the dark (300 µl/well).
- Wash 2 x with PBS.
- Incubate with Hoechst 33342 (1:4,000 in PBS) for 10 min at room temperature in the dark.
- Wash 2 x with PBS.
- Dip-wash in ddH2O.
- Mount the coverslips onto microscope slides with fluorescence mounting medium and air dry in the dark overnight.
- Store at 4 °C.
- Image using a fluorescence microscope.
- Dilute the single cell suspension obtained from one mouse in 20 ml of neurosphere growth medium containing 20 ng/ml FGF2, 20 ng/ml EGF and 2 µg/ml Heparin.
- Cell culture volumes and densities (Table 2)
Table 2. Cell culture volumes and densities
Data analysis
- In the neurosphere assay, the size and number of neurospheres can be determined and used as an indirect measure of the proliferation potential of precursor cells, for example under the influence of different external stimuli or treatments, or between differing genetic backgrounds. The neurospheres further allow the analysis of the differentiation potential of the sphere-forming cells. This can be quantified as the percentage of Map2ab/β-III-tubulin and GFAP positive cells of the total number of cells determined by Hoechst 3342 (DAPI) staining. The neurosphere assay does not allow conclusions about absolute stem cell numbers.
- In addition, to biological replications, we advise to perform neurosphere assay experiments at least five times as technical replicates since there might be some day to day differences and we always strongly advise to perform control and treatment experiments at the same time to minimize the possibility of a batch effect.
- Data analysis is performed using a Student’s t-test or an ANOVA in combination with the appropriate post-hoc test, depending on the specific experimental setup.
- Monolayer culture-based experiments allow the assessment of characteristics on a single cell level. BrdU data serve as an indirect measure of the proliferation potential of the cells by evaluating the number of cells that went through S-Phase while BrdU was present. For the differentiation experiments the number of Map2ab/β-III-tubulin positive cells and GFAP positive cells are analyzed as the percentage of the total number of cells determined by Hochest 33342 (DAPI) staining. We advise to perform both stainings simultaneously on the same coverslip for comparable results. For one differentiation experiment, we analyze four coverslips and take at least five images of each coverslip on random positions to count the cells.
- Note that freezing/thawing cycles and passaging events may influence the cells and the resulting data. It is therefore crucial to perform experiments in the same passage and with cells that have been treated similarly, to obtain reproducible data. We recommend to perform at least five technical replicates. Data analysis is performed using a Student’s t-test or an ANOVA in combination with the appropriate post-hoc test, depending on the experimental setup.
Recipes
- Trypsin inhibitor containing DNase I
0.125 mg/ml trypsin inhibitor
0.01 mg/ml DNase I
in DMEM/F-12 without glutamine - Growth medium
Neural basal medium
0.5% B-27® supplement (50x)
0.25% Pen/Strep 100,000 U/ml
0.25% GlutaMAXTM (100x stock) - Freezing medium
Growth medium
20% DMSO - Borate buffer
Boric acid
ddH2O
10 N NaOH to adjust pH to 8.5 - Antibody solution
1x PBS
3% normal donkey serum - Blocking solution
1x PBS
10% normal donkey serum
Acknowledgments
The authors declare no competing financial interests. This work was partly funded by the Deutsche Forschungsgemeinschaft SFB655 and the Bundesministerium für Bildung und Forschung. Images were acquired and processed using equipment of the Imaging Platform at the DZNE Dresden. We thank Dr. Fanny Ehret for her helpful comments on the manuscript. The protocols described here represent a variation and further development of protocols described in Babu et al. (2011), Walker and Kempermann (2014) and Ehret et al. (2016).
References
- Azari, H., Rahman, M., Sharififar, S. and Reynolds, B. A. (2010). Isolation and expansion of the adult mouse neural stem cells using the neurosphere assay. J Vis Exp (45).
- Babu, H., Claasen, J. H., Kannan, S., Runker, A. E., Palmer, T. and Kempermann, G. (2011). A protocol for isolation and enriched monolayer cultivation of neural precursor cells from mouse dentate gyrus. Front Neurosci 5: 89.
- Ehret, F., Vogler, S. and Kempermann, G. (2016). Neurosphere co-culture assay. Bio Protoc e1883.
- Hagihara, H., Toyama, K., Yamasaki, N. and Miyakawa, T. (2009). Dissection of hippocampal dentate gyrus from adult mouse. J Vis Exp (33).
- Hörster, H., Garthe, A., Walker, T. L., Ichwan, M., Steiner, B., Khan, M. A., Lie, D. C., Nicola, Z., Ramirez-Rodriguez, G. and Kempermann, G. (2017). p27kip1 is required for functionally relevant adult hippocampal neurogenesis in mice. Stem Cells 35(3):787-799.
- Palmer, T. D., Ray, J. and Gage, F. H. (1995). FGF-2 responsive neuronal progenitors reside in proliferative and quiescent regions of the adult rodent brain. Mol Cell Neurosci (5):474-86.
- Ray, J., Raymond, H. K. and Gage, F. H. (1995). Generation and culturing of precursor cells and neuroblasts from embryonic and adult central nervous system. Methods Enzymol 254:20-37.
- Reynolds, B. A. and Weiss, S. (1992). Generation of neurons and astrocytes from isolated cells of the adult mammalian central nervous system. Science 255(5052):1707-10.
- Reynolds, B. A. and Weiss, S. (1996). Clonal and population analyses demonstrate that an EGF-responsive mammalian embryonic CNS precursor is a stem cell. Dev Biol 175(1):1-13.
- Walker, T. L. and Kempermann, G. (2014). One mouse, two cultures: isolation and culture of adult neural stem cells from the two neurogenic zones of individual mice. J Vis Exp (84): e51225.
Article Information
Copyright
© 2017 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Bernas, S. N., Leiter, O., Walker, T. L. and Kempermann, G. (2017). Isolation, Culture and Differentiation of Adult Hippocampal Precursor Cells. Bio-protocol 7(21): e2603. DOI: 10.21769/BioProtoc.2603.
Category
Neuroscience > Cellular mechanisms > Cell isolation and culture
Stem Cell > Adult stem cell > Neural stem cell
Cell Biology > Cell isolation and culture > Cell differentiation
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.