- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Protocol for Construction of a Tunable CRISPR Interference (tCRISPRi) Strain for Escherichia coli
Published: Vol 7, Iss 19, Oct 5, 2017 DOI: 10.21769/BioProtoc.2574 Views: 10846
Reviewed by: Anonymous reviewer(s)
Original research article
The authors used this protocol in:

Jul 2016
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
We present a protocol for construction of tunable CRISPR interference (tCRISPRi) strains for Escherichia coli. The tCRISPRi system alleviates most of the known problems of plasmid-based expression methods, and can be immediately used to construct libraries of sgRNAs that can complement the Keio collection by targeting both essential and nonessential genes. Most importantly from a practical perspective, construction of tCRISPRi to target a new gene requires only one-step oligo recombineering. Additional advantages of tCRISPRi over other existing CRISPRi methods include: (1) tCRISPRi shows significantly less than 10% leaky repression; (2) tCRISPRi uses a tunable arabinose operon promoter and modifications in transporter genes to allow a wide dynamic range with graded control by arabinose inducer; (3) tCRISPRi is plasmid free and the entire system is integrated into the chromosome; (4) tCRISPRi strains show desirable physiological properties.
Keywords: CRISPR interferenceBackground
Various CRISPR interference systems have been developed for use in organisms from bacteria to eukaryotes. For those who are considering to use CRISPRi for bacteria, we provide the following background information on our tCRISPRi system (Li et al., 2016) and its comparison with other CRISPRi systems.
Morgan-Kiss et al. (2002) developed the plasmid-based, dose-inducible promoter pBAD. Their system allows tunable expression of a protein from the pBAD promoter, dependent upon arabinose levels. The arabinose transporter genes araE and araFGH are inactive in the strain. Their strain also has two copies of lacY; the wild-type lacY on the chromosome and a mutant lactose transporter lacY A177C on a plasmid. The LacY A177C function allows arabinose to diffuse into the cell, and thus, the pBAD induction level is precisely controlled by the concentration of the supplied arabinose in the medium (Morgan-Kiss, 2002).
Our tCRISPRi strain contains only the mutant gene lacY A177C (Morgan-Kiss, 2002), which is expressed from the lac operon constitutively because the lacI repressor gene is deleted. Our strain also has gene deletions of araE and araFGH. LacY A177C is the only arabinose transporter in the cell allowing for better control of the PBAD promoter and tunable repression by tCRISPRi.
A recent study by Peters et al. (2016) showed the power of CRISPR-based knockdown methods for studying essential genes in Bacillus subtilis. Their sgRNA libraries were cloned via inverse PCR, and dCas9 was under a xylose-inducible promoter. In contrast, our tCRISPRi system for E. coli uses one-step recombineering to make a tCRISPRi strain. The PBAD promoter in the present work shows about 7.5% leaky expression, whereas the B. subtilis CRISPRi shows approximately 33% leakiness. Another important pioneering CRISPRi system was designed by the Marraffini group (2013), who used a plasmid-based system. We compare our tCRISPRi with these other two systems in Table 1. To see an example of applications of tCRISPRi to essential cell cycle genes, see Si et al. (2017).
Table 1. Comparison of different CRISPR interference system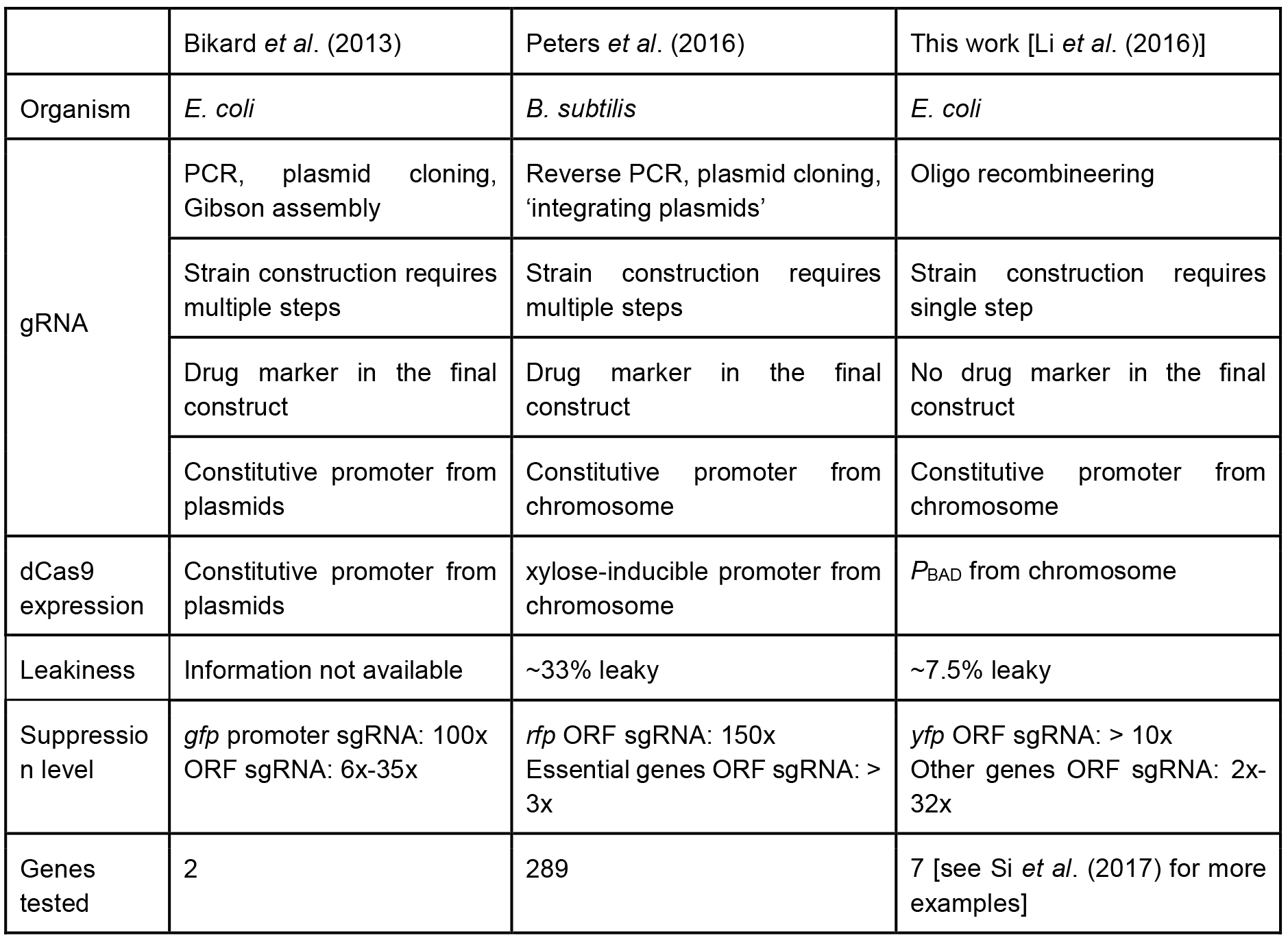
Materials and Reagents
- 1.5 ml microcentrifuge tubes
- Pipette tips (10 μl , 200 μl, 1,000 μl) (Genesee Scientific, catalog numbers: 24-121RL )
- Pipette tips (10 μl , 200 μl, 1,000 μl) (Genesee Scientific, catalog numbers: 24-150RL )
- Pipette tips (10 μl , 200 μl, 1,000 μl) (Genesee Scientific, catalog numbers: 24-165RL )
- 15 ml culture tubes
- Electroporation cuvettes, 1 mm gap (Bio-Rad Laboratories, catalog number: 1652089 )
- Petri dishes
- SJ_XTL219 strain available from Addgene (Addgene, catalog number: 86400 )
- Ultrapure water
- Antibiotics:
Hygromycin (50 mg/ml) (Thermo Fisher Scientific, InvitrogenTM, catalog number: 10687010 )
Tetracycline (12.5 mg/ml) (Sigma-Aldrich, catalog number: T3383 ) - PCR purification kit (QIAGEN, catalog number: 28104 )
- Phusion high fidelity polymerase (New England Biolabs, catalog number: M0530L )
- L(+)Arabinose (EMD Millipore, Calbiochem, catalog number: 178680 )
- Gel extraction kit (QIAGEN, catalog number: 28704 )
- Tryptone (EMD Millipore, catalog number: 1.07213.1000 )
- Yeast extract (Sigma-Aldrich, catalog number: Y1625-1KG )
- Sodium chloride (NaCl) (Fisher Bioreagents, catalog number: BP3581 )
- LB agar (Fisher Bioreagents, catalog number: BP1425 )
- Luria broth (LB) (see Recipes)
- LB agar plate (see Recipes)
- Sucrose plates (see Recipes)
Equipment
- Pipettes (that can accommodate pipette tips in 2 above)
- Milli-Q water purification system (Millipore, catalog number: Z00Q0V0WW )
- Autoclave (AMSCO, model: 3041-S )
- Incubator and shaker (32 °C and 42 °C) (Eppendorf, New BrunswickTM, model: Innova® 3100 , catalog number: M1231-0000)
- Microcentrifuge (Eppendorf, catalog number: 5424 )
- Electroporator (Bio-Rad Laboratories, catalog number: 1652100 )
- Gel electrophoresis chamber (Bio-Rad Laboratories, catalog number: 1704406 )
- Thermal cycler (Bio-Rad Laboratories, model: C1000 TouchTM, catalog number: 1851148 )
- Nikon Inverted Microscope Eclipse Ti-E (Nikon, model: Eclipse Ti-E , catalog number: MEA53100) equipped with an Andor Neo sCMOS camera (Nikon, model: Neo 5.5 , catalog number: 77026046)
Software
- Nikon NIS-Elements Advanced Research software
Procedure
- Design sgRNA targeting your gene of interest. Oligos were designed for every potential sgRNA sequence, see Li et al. (2016).
- Assemble oligo
a.Left homology arm to SJ_XTL219 (TTGACAGCTAGCTCAGTCCTAGGTATAATGCTAGC).
b.Reverse compliment of target sequence without ngg.
c.Right homology arm to SJ_XTL219 (GTTTTAGAGCTAGAAATAGCAAGTTAAAATAAG). - Order completed Standard Desalting oligos from an Oligonucleotide synthesis company.
- Recombine sgRNA into SJ_XTL219
- Grow SJ_XTL219 in liquid LB culture (see Recipes) supplemented with hygromycin (75 μg/ml) in a 32 °C shaker for 2.5-3 h until OD600 between 0.3-0.6. The recombineering plasmid pSIM18 has a temperature sensitive replication origin, so be sure to keep your culture at 32 °C. The volume of culture depends on the number of recombinations you are performing. We recommend 5 ml of culture per reaction. See Chan et al. (2007) for more information.
- Heat shock the culture in a 42 °C shaker for 15 min. This activates lambda RED recombination system in pSIM18, which is already in SJ_XTL219.
- Transfer culture immediately to ice water for at least 10 min, 3 h in the ice water will not affect the recombineering frequency.
- Centrifuge the culture to form a pellet at 6,300 x g for 7 min at 4 °C by using refrigerated, low-speed centrifuge with Sorvall SA-600 rotor (or equivalent).
- Discard the supernatant.
- Resuspend the pellet with 30 ml of cold ultrapure water by pipetting up and down.
- Centrifuge again at 6,300 x g for 7 min at 4 °C by using refrigerated, low-speed centrifuge with Sorvall SA-600 rotor (or equivalent).
- Discard the supernatant by inversion.
- Resuspend the pellet in 1 ml of cold ultrapure water.
- Transfer the cells to a 1.5 ml Eppendorf tube and centrifuge at maximum speed 30 sec.
- Discard the supernatant.
- Resuspend the cells in cold ultrapure water by pipetting up and down. For each electroporation, 50 μl resuspend cells are needed.
- Add 1 μl of your oligo (10 picomole/μl) to 50 μl competent cells and mix by pipetting up and down.
- Transfer the mixture into a chilled electroporation cuvette. Aliquot 50 μl of competent cells to a chilled electroporation cuvette as the no oligo control.
- Electroporation. Choose the ‘Bacteria’ setting.
- Add 1 ml of LB to the electroporation cuvette and mix with the cells with the pipette to draw out the cells.
- Transfer to a culture tube with 10 ml room temperature LB broth and shake in a 32 °C water bath shaker for 4 h.
- Spread the cell culture onto sucrose plates (see Recipes) after 4 h outgrowth in the 32 °C water bath.
- Place plates in a 32 °C incubator overnight.
- Grow SJ_XTL219 in liquid LB culture (see Recipes) supplemented with hygromycin (75 μg/ml) in a 32 °C shaker for 2.5-3 h until OD600 between 0.3-0.6. The recombineering plasmid pSIM18 has a temperature sensitive replication origin, so be sure to keep your culture at 32 °C. The volume of culture depends on the number of recombinations you are performing. We recommend 5 ml of culture per reaction. See Chan et al. (2007) for more information.
- Check colonies for successful insertion through colony PCR
- Use primers:
- Sense primer: TCCTGCGTCCTGGCGAAGAG
- Anti-sense primer: CCATGACGAACCAGAACCAG
- Sense primer: TCCTGCGTCCTGGCGAAGAG
- The correct product should be 842 bp.
- Sequencing for further confirmation by using one of the above checking PCR primers (we use a sequencing service).
- Use primers:
- Transcriptional reporter (as a way of monitoring how much your gene of interest is being down expressed)
- Make a strain that has a fluorescent gene with an antibiotic marker nearby that is not hygromycin (Li et al., 2016). Design primers to PCR those elements. Make the homologous ends of the primers match the sequence before your gene of interest. The primer to amplify the fluorescent gene must have a Shine-Dalgarno sequence (Li et al., 2016). We can provide a strain where the chloramphenicol resistant gene is located downstream of the msfGFP (SJ_XTL404) (Li et al., 2016).
- PCR the strain SJ_XTL404 with your designed primers and purify the product from gel using a kit.
- Recombine the fluorescent gene and drug marker PCR cassette into the CRISPRi strain which is confirmed in step 5.
- After the recombineering, outgrow the cells at 32 °C for more than 2 h.
- Plating the cells on suitable antibiotic plates, grow in a 32 °C incubator overnight.
- Colony PCR to check the recombinants.
- Make a strain that has a fluorescent gene with an antibiotic marker nearby that is not hygromycin (Li et al., 2016). Design primers to PCR those elements. Make the homologous ends of the primers match the sequence before your gene of interest. The primer to amplify the fluorescent gene must have a Shine-Dalgarno sequence (Li et al., 2016). We can provide a strain where the chloramphenicol resistant gene is located downstream of the msfGFP (SJ_XTL404) (Li et al., 2016).
- Induction with arabinose
- Start seed culture in LB overnight.
- Back dilute 2 μl overnight culture in 2 ml of your target media (can be LB or any media you need for your experiment).
- When the first back dilution reaches exponential phase after around 2 h (~OD600 = 0.1) back dilute 5 μl culture to 955 μl media and grow at 37 °C for about 3.5 h. We recommend making multiple culture tubes with this back dilution to test different arabinose concentrations.
- Add 10% arabinose to your back dilution. How much you add depends on the level of down expression you want. We recommend making a gradient to test what concentrations work well for you.
- If imaging, fix your cells when cultures are turbid.
- Start seed culture in LB overnight.
- Examples of knockdown of essential and nonessential genes using tCRISPRi
We presented several examples of gene repression by tCRISPRi in Figure 1. They are three essential genes rpoB, dnaG, and ftsZ, and three nonessential genes mCherry, lexA and lacZ. To quantify the level of knockdown by tCRISPRi, we employed a msfGFP fluorescent transcription reporter system.
For these six genes, we observed as much as 32-fold inhibition, with several genes in the range of 2- to 3-fold inhibition. More specifically, for all three essential genes rpoB, dnaG, ftsZ, we observed up to 14-fold knockdown in gene expression. The knockdown of these genes caused significant increase in cell size (Figure 1C). For the non-essential genes mCherry, lexA and lacZ, the expression level decreased up to 32-fold, and neither the growth rates nor the cell size were affected by the knockdown (Figure 1C).
These results show that the tCRISPRi system can be used for graded suppression of gene expression for both essential and nonessential genes from their wild-type level with minimal leaky expression (Figure 1).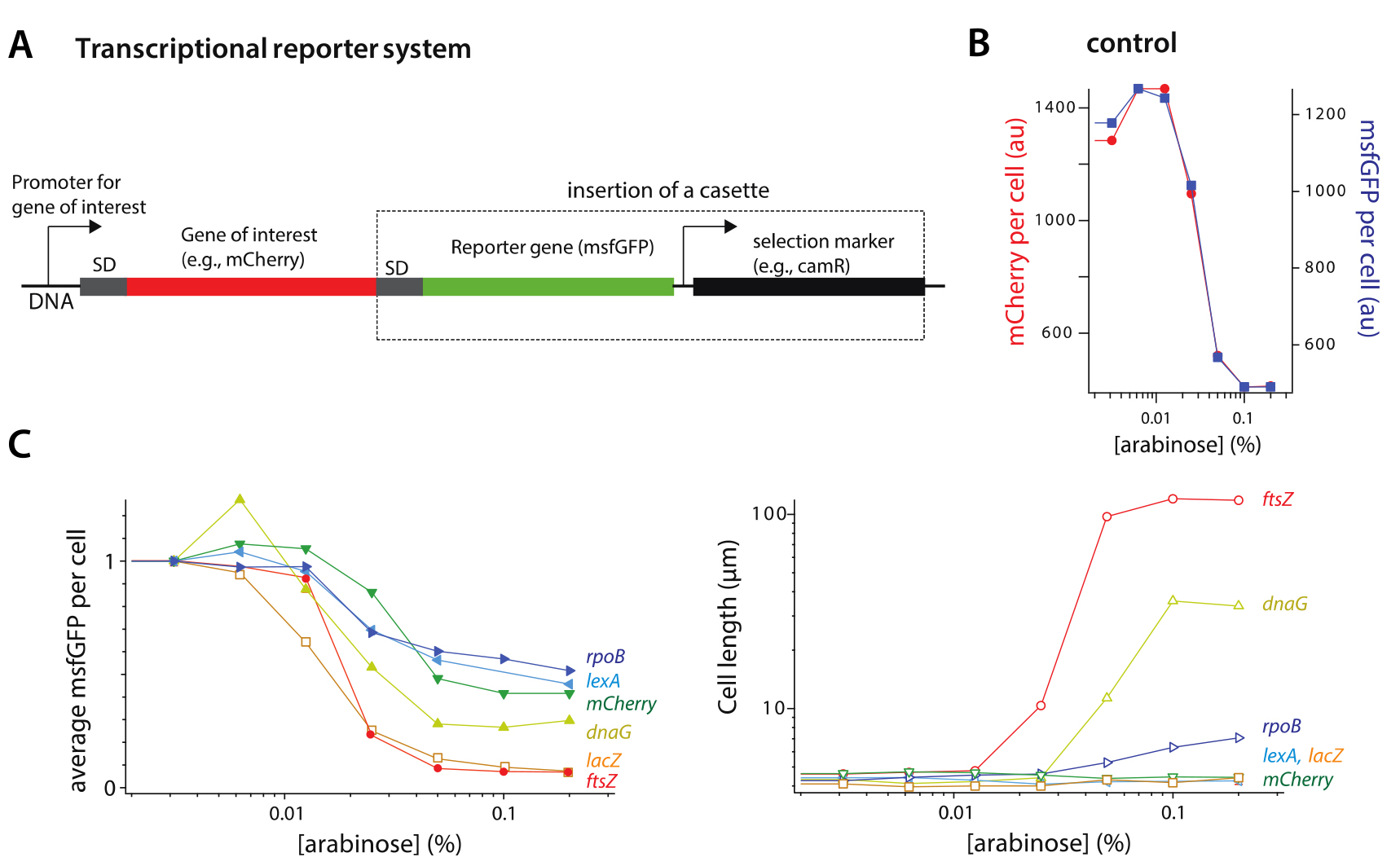
Figure 1. Application of tCRISPRi to essential and nonessential genes. A. Design of the fluorescent transcriptional reporter. B. Control of the transcriptional reporter (msfGFP). mCherry is the gene of interest, and its fluorescence level changes as the same rate as the transcriptional reporter (msfGFP) under knockdown of mCherry by tCRISPRi. C. Knockdown of ftsZ, dnaG, and rpoB (essential genes) and lacZ, lexA, mCherry (nonessential genes). The expression level of all six genes decreased by tCRISPRi from their wildtype level up to 26 folds. Cell length of the three essential genes all increased by knockdown, whereas cell length remained unchanged for the nonessential genes.
Data analysis
Images were acquired at 100x magnifications using Nikon Ti-E microscope equipped with a Neo sCMOS camera (Andor) and Nikon NIS-Elements software. For the cell length measurements, phase contrast technique was used, and images have 2,560 x 2,160 resolution and 16 bit grayscale. The fluorescent images for msfGFP cells were obtained with illumination by an OBIS 488 nm laser from Coherent and the 59022 filter cube from Chroma. For each cell, the fluorescence signal was integrated and normalized by the projected area of the cell after background subtraction. Illumination across the field of view was homogeneous with less than 5% variations. For each experimental condition, we acquired 150-200 images containing at least 10,000 cells, and calculated the average value of each steady-state population for the data in Figure 1. We developed and used custom high-throughput image analysis software optimized for our experiments using Python and OpenCV library. For more detailed information on data analysis is available in Materials and Methods as well as Supplementary Information in Li et al., 2016 with specific examples of MreB, DnaA, RepA, SeqA knockdown in Si et al., 2017.
Recipes
- Luria broth (LB) for 400 ml
4 g tryptone
2 g yeast extract
2 g NaCl
Add ultrapure water up to 400 ml
Autoclave
Store at room temperature - LB agar plate for 400 ml
4 g tryptone
2 g yeast extract
2 g NaCl
4.8 g LB agar
Add ultrapure water up to 400 ml
Autoclave
Pour into Petri dishes
Let cool completely and store at 4 °C - Sucrose plates for 800 ml
54 g sucrose dissolved completely in water
9 g tryptone
4.5 g yeast extract
10.8 g LB agar
Add ultrapure water up to 800 ml
Autoclave
Pour into Petri dishes
Let cool completely and store at 4 °C
Acknowledgments
We thank Don Court (NCI/NIH) for collaboration with the published work in Li et al. (2016). This work was supported by the Paul G. Allen Foundation, the Pew Charitable Trusts, the National Science Foundation CAREER Award, and NIH R01 GM118565-01 (to S.J.). We thank Sarah E. Cox for critical reading of the manuscript.
References
- Bikard, D., Jiang, W., Samai, P., Hochschild, A., Zhang, F. and Marraffini, L. A. (2013). Programmable repression and activation of bacterial gene expression using an engineered CRISPR-Cas system. Nucleic Acids Res 41(15): 7429-7437.
- Chan, W., Costantino, N., Li, R., Lee, S. C., Su, Q., Melvin, D., Court, D. L. and Liu, P. (2007). A recombineering based approach for high-throughput conditional knockout targeting vector construction. Nucleic Acids Res 35(8): e64.
- Li, X. T., Jun, Y., Erickstad, M. J., Brown, S. D., Parks, A., Court, D. L. and Jun, S. (2016). tCRISPRi: tunable and reversible, one-step control of gene expression. Sci Rep 6: 39076.
- Morgan-Kiss, R. M., Wadler, C. and Cronan, J. E., Jr. (2002). Long-term and homogeneous regulation of the Escherichia coli araBAD promoter by use of a lactose transporter of relaxed specificity. Proc Natl Acad Sci U S A 99(11): 7373-7377.
- Peters, J. M., Colavin, A., Shi, H., Czarny, T. L., Larson, M. H., Wong, S., Hawkins, J. S., Lu, C. H., Koo, B. M., Marta, E., Shiver, A. L., Whitehead, E. H., Weissman, J. S., Brown, E. D., Qi, L. S., Huang, K. C. and Gross, C. A. (2016). A comprehensive, CRISPR-based functional analysis of essential genes in bacteria. Cell 165(6): 1493-1506.
- Si, F., Li, D., Cox, S. E., Sauls, J. T., Azizi, O., Sou, C., Schwartz, A. B., Erickstad, M. J., Jun, Y., Li, X. and Jun, S. (2017). Invariance of initiation mass and predictability of cell size in Escherichia coli. Curr Biol 27(9): 1278-1287.
Article Information
Copyright
© 2017 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Li, X., Sou, C. and Jun, S. (2017). Protocol for Construction of a Tunable CRISPR Interference (tCRISPRi) Strain for Escherichia coli. Bio-protocol 7(19): e2574. DOI: 10.21769/BioProtoc.2574.
Category
Molecular Biology > DNA > DNA modification
Microbiology > Microbial genetics > Gene mapping and cloning
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.