- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

An Optimized Method for the Production Using PEI, Titration and Neutralization of SARS-CoV Spike Luciferase Pseudotypes

Published: Vol 7, Iss 16, Aug 20, 2017 DOI: 10.21769/BioProtoc.2514 Views: 17075
Reviewed by: Longping Victor TseSmita NairDavid Paul
Original research article
The authors used this protocol in:

Mar 2005
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
The protocol outlined represents a cost-effective, rapid and reliable method for the generation of high-titre viral pseudotype particles with the wild-type SARS-CoV spike protein on a lentiviral vector core using the widely available transfection reagent PEI. This protocol is optimized for transfection in 6-well plates; however it can be readily scaled to different production volumes according to application. This protocol has multiple benefits including the use of readily available reagents, consistent, high pseudotype virus production Relative Luminescence Units (RLU) titres and rapid generation of novel coronavirus pseudotypes for research into strain variation, tropism and immunogenicity/sero-prevalence.
Keywords: SARS coronavirusBackground
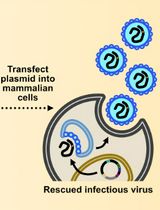
The production and use of pseudotyped viral particles (PV) is widely established for many viruses, and applications in the fields of serology, surveillance and vaccine development are manifold (Temperton et al., 2015; Carnell et al., 2015). PVs have proven to be powerful tools to study the effects of viral envelope glycoprotein mutations on serological outcomes, viral tropism and immunogenicity studies especially when combined with epitope information. PVs are chimeric viral constructs in which the outer (surface) glycoprotein(s) of one virus is combined with the replication-defective viral ‘core’ of another virus. PV allow for accurate, sequence-directed, sensitive antibody neutralization assays and antiviral screening to be conducted within a low biosecurity facility and offer a safe and efficient alternative to wildtype virus use, making them exquisitely beneficial for many emerging RNA viruses of pandemic potential. Many of the published protocols require modification of the SARS spike glycoprotein and/or expensive transfection reagents (Temperton, 2009). The protocol presented here utilizes the full-length, non-codon-optimized spike protein in conjunction with the low-cost transfection reagent PEI, making this protocol widely applicable to many stakeholder laboratories. Figure 1 shows a cartoon of the lentiviral SARS-CoV PV production process directed by plasmid co-transfection.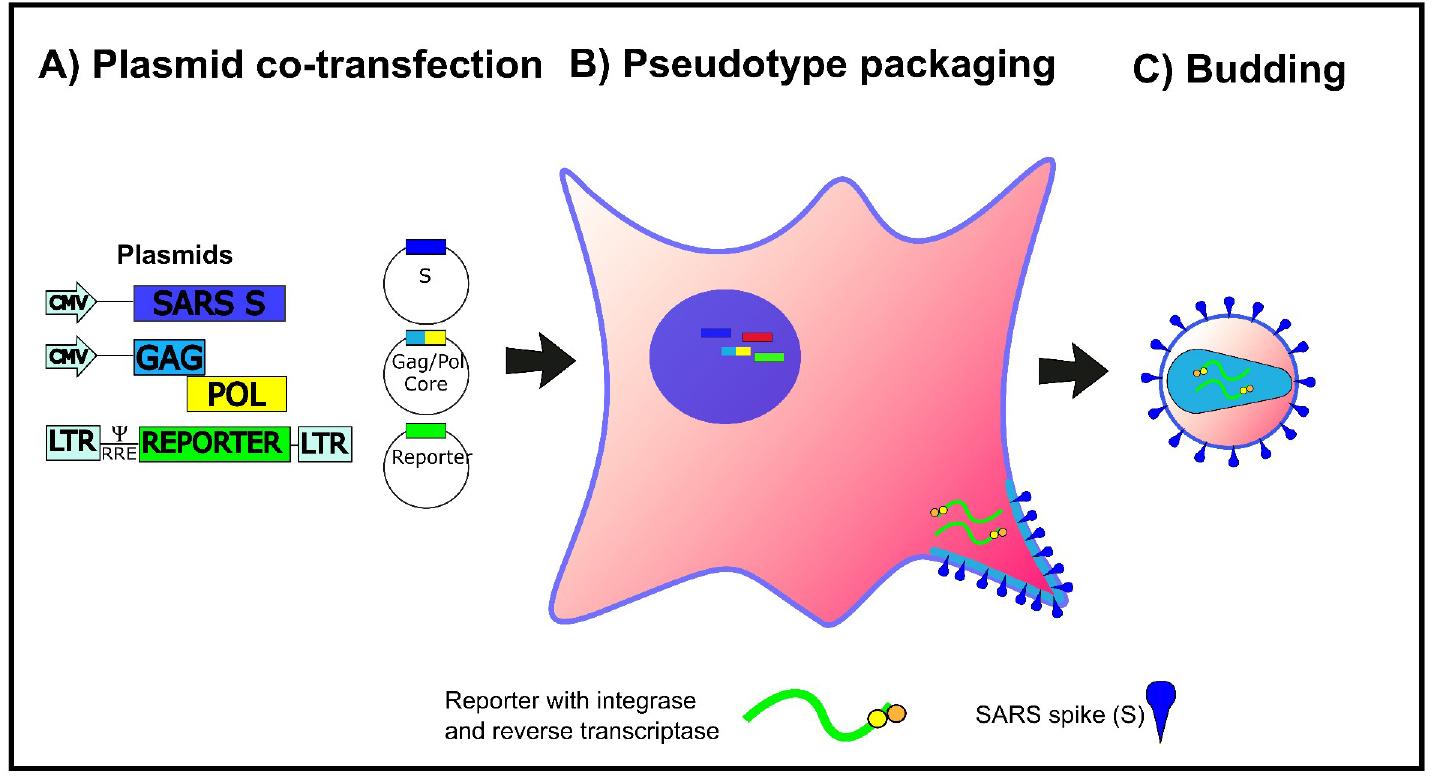
Figure 1. Cartoon representation of the production of SARS pseudotypes. HEK293T/17 cells are transfected with three plasmids, bearing the relevant genes (Lentiviral vector, packaging construct and SARS-CoV spike expression plasmid) for the production of SARS-CoV Spike bearing lentiviral pseudotypes. This figure is modified from Carnell et al. (2015).
Materials and Reagents
- MultiGuard Barrier pipette tips 1-20 and 1-200 μl (Sorenson BioScience, catalog number: 30550T )
- NuncTM Cell-Culture Treated Multidishes (6-well) (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 140675 )
- Microcentrifuge tube Safe-Lock write-on graduated with lid latch 1.5 ml
- Sterile syringes (10 ml), Generic
- Millex-HA 0.45 µm filters (Merck, catalog number: SLHAM33SS )
- 96-well white plate (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 136101 )
- HEK 293T/17 cells (ATCC, catalog number: CRL-11268 )
- Huh7 cells (Cell Signaling Technology, catalog number: 300156 )
- Plasmids
- Glycoprotein expression plasmid: pCAGGS-SARS-CoV spike
- Lentiviral vector expressing firefly luciferase: pCSFLW
- Second-generation lentiviral packaging construct plasmid: p8.91 (expresses gag, pol and rev)
Note: Information on the plasmids above can be found in Temperton et al. (2005) and Carnell et al. (2015). Plasmids available from Viral Pseudotype Unit, University of Kent. n.temperton@kent.ac.uk - Glycoprotein expression plasmid: pCAGGS-SARS-CoV spike
- Dulbecco’s modified Eagle medium (DMEM) with GlutaMAX (Thermo Fisher Scientific, catalog number: 31966021 ) supplemented with 10% foetal bovine serum (FBS) (Pan-Biotech, catalog number: P40-37500 ) and 1% penicillin/streptomycin (P/S) (Pan Biotech, catalog number: P06-07100 )
- Gibco Reduced Serum media Opti-MEM® (Thermo Fisher Scientific, catalog number: 31985047 )
- Branched Polyethyleneimine (PEI) solution at concentration of 1 mg/ml (Sigma-Aldrich, catalog number: 408727 ).
Note: PEI is dissolved in dH2O to a concentration of 1 mg/ml and the pH is adjusted to 7 using diluted (1:3) concentrated HCl. - Phosphate-buffered saline (PBS)
- Trypsin-EDTA (0.05%), phenol red (Thermo Fisher Scientific, GibcoTM, catalog number: 25300054 )
- Positive control antibody (monoclonal/polyclonal or post-infection serum) that can neutralize the SARS pseudotype
- Bright GloTM luciferase assay system (Promega, catalog number: E2650 )
Equipment
- Class II biosafety cabinet (Thermo Fisher Scientific, Thermo ScientificTM, model: MSC-AdvantageTM )
- Water bath or incubator
- Pipettes (Gilson, model: PIPETMAN® Classic, P2 , P20 , P200 and P1000 )
- Optional: BIO-RAD TC20TM Automated Cell Counter (Bio-Rad Laboratories, catalog number: 1450102EDU )
- Plate centrifuge (ELMI, model: SkyLine CM-6MT )
- Glomax 96 luminometer (Promega, model: GloMax® 96 )
Note: This product has been discontinued.
Procedure
- Transfection steps
Note: All steps should be carried out in a class II biosafety cabinet to avoid contamination. Timeline: Transfection–24 h.- 293T/17 cells should be subcultured into 6-well plates at a ratio that will deliver 70-90% confluence at the time of transfection. Typically seeding 4 x 105 cells per well will achieve this level of confluency. Example is shown in Figure 2.
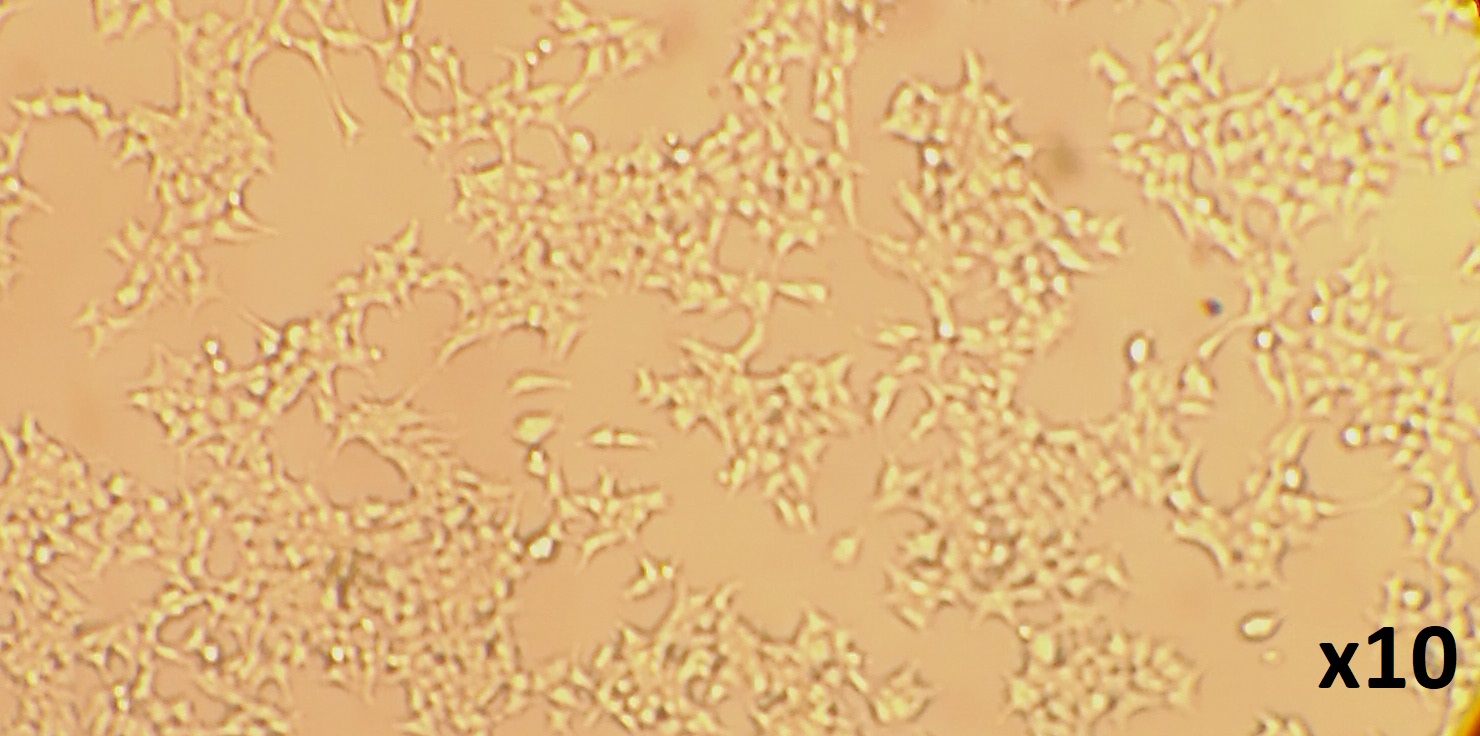
Figure 2. Example of the confluency expected prior to transfection of HEK293T/17 cells
- DMEM/10% FBS/1% P/S and Opti-MEM® should be pre-warmed to 37 °C using a water bath or an incubator.
- Prepare two labelled sterile 1.5 ml microcentrifuge tubes (tube 1 and tube 2) for each well of a 6-well plate which will be used for transfections.
- Add the following plasmid constructs for transfection to tube 1:
pCAGGS-SARS-CoV spike: 450 ng
p8.91-lentiviral vector: 500 ng
pCSFLW: 750 ng - Add 100 µl Opti-MEM® to the plasmid DNA mix (tube 1).
- Add 100 µl Opti-MEM® and 17.5 µl of 1 mg/ml PEI to tube 2.
- Incubation step: Mix both tubes by gently flicking and incubate for 5 min at room temperature (RT).
- After incubation, pipette the Opti-MEM®/PEI solution from tube 2 into the Opti-MEM®/DNA solution in tube 1.
- Incubation step: Incubate the tube at RT for 20 min whilst gently flicking the tube to mix every 3-4 min.
- Whilst the transfection mix is incubating, the culture media on the 293T/17 cells should be removed and 2 ml of fresh DMEM/10% FBS/1% P/S added per well. It is imperative at this point to add culture media slowly to one side of the well to avoid detaching the adherent cell monolayer.
- After 20 min incubation, pipette the DNA/Opti-MEM®/PEI solution onto the 293T/17 cells by adding dropwise throughout the total surface area of the well. Swirl the 6-well plate(s) gently to ensure an even dispersal of reagent mix.
- Incubation step: Incubate the plate at 37 °C, 5% CO2 overnight (o/n). In our hands incubation times of between 12-16 h result in equivalent final PV production RLU titres.
- Post o/n incubation the culture media on the cells should be carefully removed and 2 ml fresh DMEM/10% FBS/1% P/S added. Again, add media slowly to one side of the well to avoid cell detachment.
- Incubate the 6-well plates at 37 °C 5% CO2 o/n for 32-36 h.
- Supernatant containing the viral pseudotype particles are harvested using a 2.5 ml sterile syringe and subsequently filtered into Falcon or microcentrifuge tubes via a syringe driven Millex HA-0.45 µm filter.
- Store all filtered supernatants at -80 °C until downstream use. It is recommended that supernatant is stored as aliquots to avoid multiple freeze-thaw cycles that may impact viral RLU titres.
Note: Supernatant may be stored at 4 °C for up to one week with no detectable loss of RLU titre. - Optional step: Additional culture media may be added to cells to allow a second harvest 18-24 h later by adding further DMEM/10% FBS/1% P/S. In this case, extreme care must be taken in initial PV collection (step A15) to avoid damage to the cell monolayer. We have observed that cells in poor health after first harvest result in significantly lower PV production RLU titres upon second harvest.
- 293T/17 cells should be subcultured into 6-well plates at a ratio that will deliver 70-90% confluence at the time of transfection. Typically seeding 4 x 105 cells per well will achieve this level of confluency. Example is shown in Figure 2.
- Titration steps (Figure 3)
Note: Titration consists of transduction of reporter (in this case firefly luciferase) into target cells mediated by the viral glycoprotein expressed on the viral pseudotype (SARS-CoV spike). Controls for titration are provided via the inclusion of ‘cell only’ and ‘Delta Env’ columns. Control for transduction can be provided via a PV bearing the Vesicular stomatitis virus G protein (VSV-G) which utilizes a ubiquitous receptor which results in high RLU titres in all cell lines tested.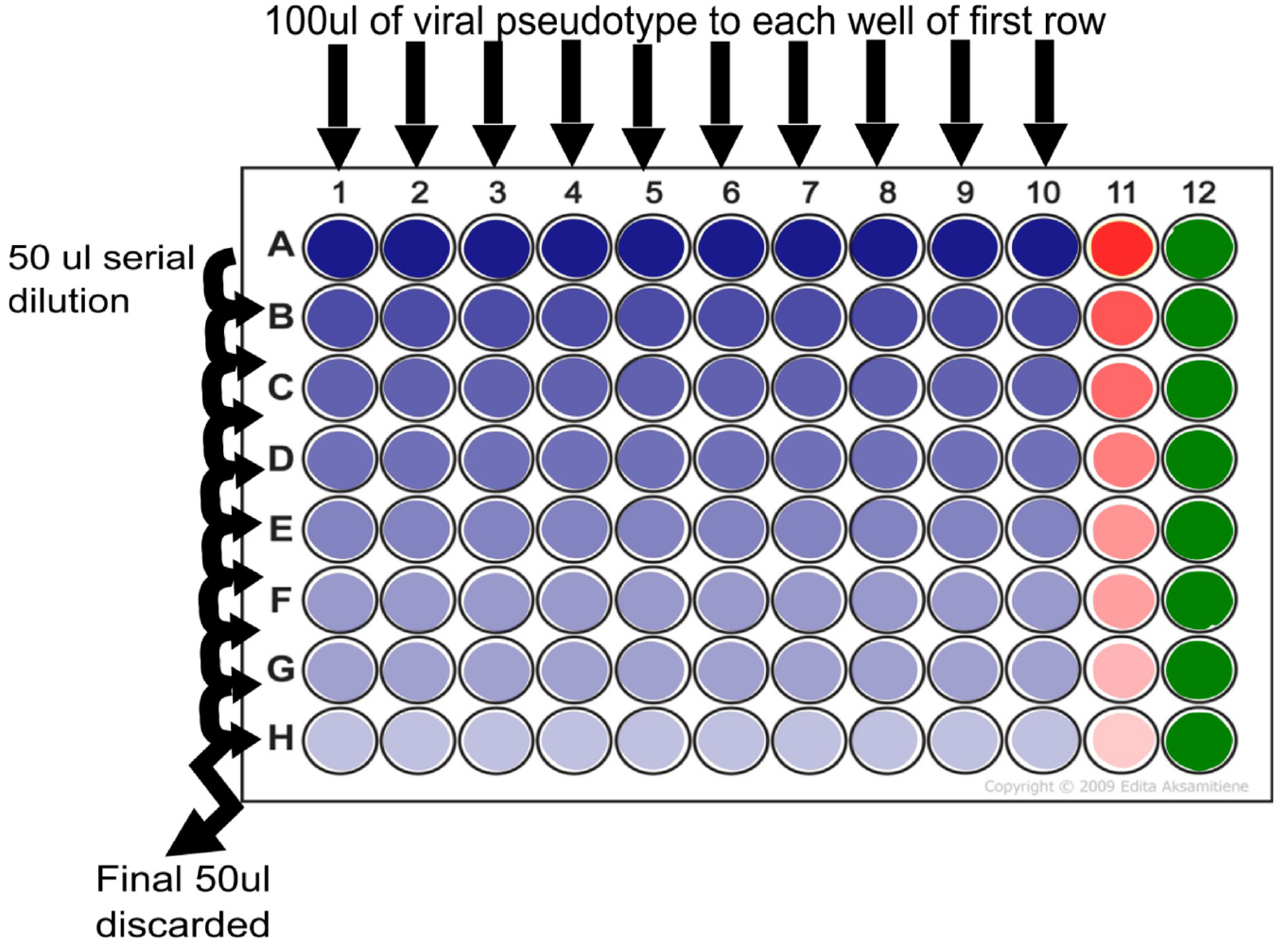
Figure 3. 96-well plate set-up for pseudotype titration. Serial dilution step showing addition of 100 µl of pseudotype virus supernatant to each well of row A and dilution of 50 µl taken from this well to row B. This process is continued to end of plate (row H) at which point the final 50 µl is discarded. Delta Env control is indicated in red (column 11) and cell only control is indicated in green (column 12). One set of pipette tips can be used per dilution series (plate).- In a 96-well white plate add 50 µl of DMEM/10% FBS/1% P/S to the entire column of ‘cell only’ control (see Figure 3 column 12).
- Add 50 µl of DMEM/10% FBS/1% P/S from row B to H that are to contain PV or Delta-Env control virus.
- Add 100 µl of SARS pseudotype virus supernatant to each well of row A (excluding control columns) and add 100 µl of Delta-Env to column 11 (see Figure 3).
- Remove 50 µl from row A virus-containing wells and perform two-fold serial dilutions down all the wells beneath it.
- With each dilution step use a pipette to mix 8 times by pipetting up and down and taking care not to produce air bubbles.
- After completing serial dilution the final 50 µl from the final well of each column should be discarded.
Note: At this point each well should contain 50 µl of mixed DMEM and PV supernatant. - Prepare a plate of susceptible target cells (Huh-7 for SARS PV), preferentially subcultured 1:4 48 h before:
- Remove culture media from plate.
- Wash the plate twice with 2 ml of PBS and discard.
- Add 2 ml of trypsin to the plate to detach cells.
- After cells have detached add 6 ml of DMEM/10% FBS/1% P/S to the plate to quench trypsin activity, and resuspend cells gently.
- Count cells using TC20TM Automated Cell Counter or haemocytometer and add 1 x 104 cells in a total volume of 50 µl to each well.
- Remove culture media from plate.
- Centrifuge plate for 1 min at 800 x g (Rcf) if there are visible droplets on the sides of the wells.
- Incubate the plate for 48 h at 37 °C 5% CO2.
- Read plate using Bright GloTM luciferase assay system on a Glomax 96 luminometer (or equivalent).
- In a 96-well white plate add 50 µl of DMEM/10% FBS/1% P/S to the entire column of ‘cell only’ control (see Figure 3 column 12).
- Pseudotype based neutralisation assay (pMN)
Note: pMN is the Inhibition of PV mediated transduction via an antibody (or inhibitor) directed against the SARS glycoprotein.- In a 96-well white plate add 50 µl of DMEM/10% FBS/1% P/S to rows B to H, columns 1-12.
- Add known amount of antibody (example 5 µl HN and R sera in Figure 4) into wells of row A, columns 2-10 in a total volume of 100 µl DMEM/10% FBS/1% P/S. Add known amount (e.g., 5 µl) of positive and negative antisera into wells A11 and A12 as controls.
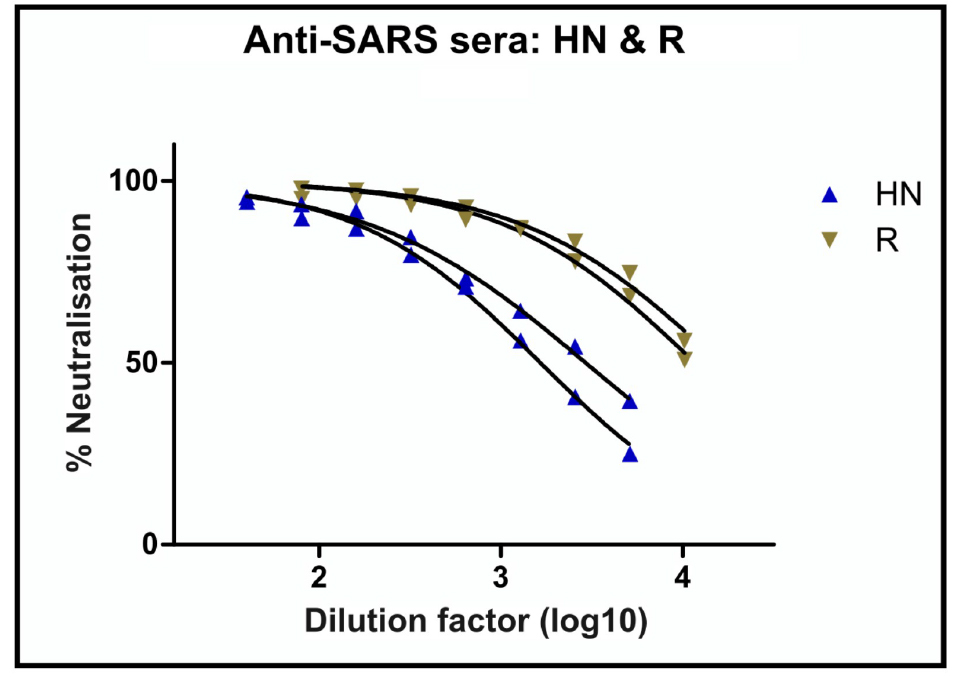
Figure 4. Anti-SARS antibodies (post-infection sera HN and R) neutralization of SARS viral pseudotype entry into Huh7 cells. Two repeats are plotted for each serum sample. - Remove 50 µl from row A wells and perform two-fold serial dilutions down all the wells beneath it.
- With each dilution step use a pipette to mix 15 times by pipetting up and down and taking care not to produce air bubbles.
- After completing serial dilution the final 50 µl from the final well of each column should be discarded.
Note: At this point each well should contain 50 µl of mixed DMEM and serial dilutions of antibody/inhibitor. - Centrifuge plate for 1 min at 800 x g (Rcf) to ensure no inhibitor or liquid is located on the walls of the well.
- Using data obtained from the titration, calculate the amount of DMEM required to dilute your SARS-PV to obtain 1 x 106 RLU in 50 µl, with a total volume of 5 ml. For example with an RLU/ml of 1 x 108, 1 ml of PV should be mixed with 4 ml of DMEM.
- Mix this diluted PV solution using the multichannel pipette, and aliquot 50 µl into each well on the plate, with the exception of wells A6-A12 (cell only control). A1-A6 will serve as virus only control.
- Centrifuge plate for 1 min at 800 x g (Rcf) to ensure no virus is left on the walls of the well.
- Incubate the plates for 1 h at 37 °C 5% CO2, allowing time for the antibody/inhibitor to bind the SARS glycoprotein.
- Prepare a plate of susceptible target cells (Huh-7 for SARS PV), preferentially subcultured 1:4 48 h before:
- Remove culture media from plate.
- Wash the plate twice with 2 ml of PBS and discard.
- Add 2 ml of trypsin to the plate to detach cells.
- After cells have detached add 6 ml of DMEM/10% FBS/1% P/S to the plate to quench trypsin activity, and resuspend cells gently.
- Count cells using TC20TM Automated Cell Counter or haemocytometer and add 1 x 104 cells in a total volume of 50 µl to each well.
- Remove culture media from plate.
- Incubate the plate for 48 h at 37 °C 5% CO2.
- Read plate using Bright GloTM luciferase assay system on a Glomax 96 luminometer (or equivalent).
- In a 96-well white plate add 50 µl of DMEM/10% FBS/1% P/S to rows B to H, columns 1-12.
Data analysis
- RLU readings are multiplied to RLU/ml by the dilution factor of each well (20x, 40x, 80x, 160x, 320x, 640x, 1,280x, 2,560x). The mean of all 8 RLU/ml values is used as the final value reported for that column in the titration step. A linear relationship should be observed between RLU values and PV dilution, with values decreasing by 50% after each 1:2 dilution. Care should be taken to check this linear relationship before multiplication, as this inherently can lead to false positives despite lack of PV or working PV (Table 1).
Table 1. Analysis of PV titration data. RLU values are multiplied to give an RLU/ml value (example highlighted in green) giving RLU/ml values for each of the dilution points. The mean/average is then calculated from all 8 dilution points. Care must be taken to observe a linear relationship between dilution factor (X factor) and RLU, or multiplication can lead to inflated RLU/ml values (example highlighted in yellow).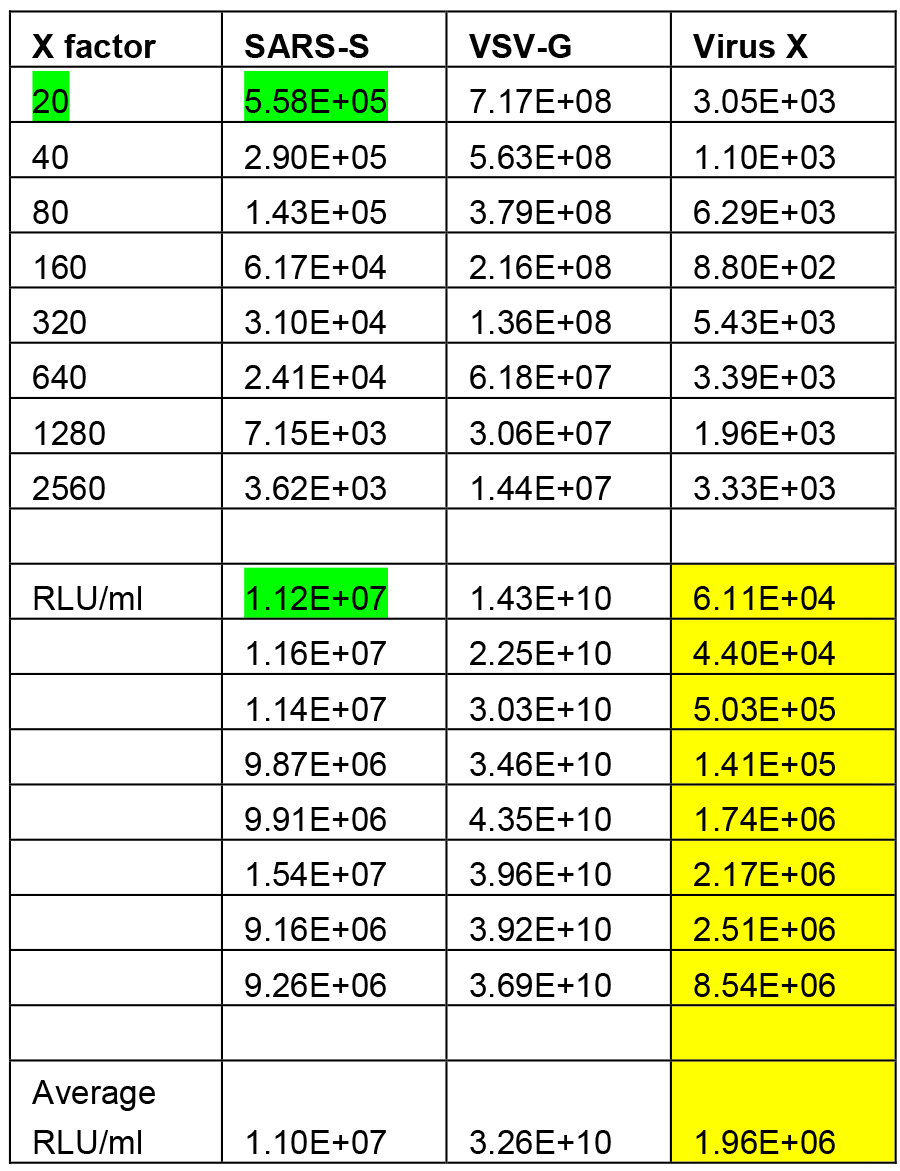
- Protocol read-out and titration results (Figure 5).
Figure 5. Results of pseudotype production RLU titres using optimized transfection protocol. SARS; PV with SARS-CoV Spike on surface, Cell; cell only control, Delta Env; PV with no surface glycoprotein, VSV-G; PV with VSV-G on surface. - The protocol outlined provides a rapid and consistent method for the generation of high-titre viral pseudotype particles expressing the SARS-CoV spike protein suitable for further downstream R&D applications. Efficient knock-down (neutralization) of SARS-CoV pseudotype virus entry using two post-infection sera (HN and R) demonstrates potential utility for vaccine immunogenicity and mAb/antiviral screening. The use of readily available reagents should facilitate increased reproducibility.
Acknowledgments
The authors acknowledge Dr Paul Chan (Faculty of Medicine, The Chinese University of Hong Kong) for the two positive post-infection control sera. This work was funded by the Medway School of Pharmacy, Kent, UK. This protocol was modified and improved from PMID: 15757556 and 26587388.
References
- Carnell, G. W., Ferrara, F., Grehan, K., Thompson, C. P. and Temperton, N. J. (2015). Pseudotype-based neutralization assays for influenza: a systematic analysis. Front Immunol 6: 161.
- Grehan, K., Ferrara, F. and Temperton, N. (2015). An optimised method for the production of MERS-CoV spike expressing viral pseudotypes. MethodsX 13(2): 379-384.
- Temperton, N. J. (2009). The use of retroviral pseudotypes for the measurement of antibody responses to SARS coronavirus. In Lal, S. K. (Ed.). Molecular biology of the SARS-coronavirus. Springer.
- Temperton, N. J., Chan, P. K., Simmons, G., Zambon, M. C., Tedder, R. S., Takeuchi, Y. and Weiss, R. A. (2005). Longitudinally profiling neutralizing antibody response to SARS coronavirus with pseudotypes. Emerg Infect Dis 11(3): 411-416.
- Temperton, N. J., Wright, E. and Scott, S. D. (2015). Retroviral pseudotypes–from scientific tools to clinical utility. eLS 1-11.
Article Information
Copyright
© 2017 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Carnell, G., Grehan, K., Ferrara, F., Molesti, E. and Temperton, N. (2017). An Optimized Method for the Production Using PEI, Titration and Neutralization of SARS-CoV Spike Luciferase Pseudotypes. Bio-protocol 7(16): e2514. DOI: 10.21769/BioProtoc.2514.
Category
Microbiology > Microbe-host interactions > Virus
Microbiology > Microbe-host interactions > Pseudovirus
Biochemistry > Protein > Synthesis
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.