- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Advanced Design of Minimalistic Dumbbell-shaped Gene Expression Vectors


Published: Vol 7, Iss 15, Aug 5, 2017 DOI: 10.21769/BioProtoc.2425 Views: 10446
Reviewed by: Longping Victor TseKanika GeraAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Sep 2016
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
Minimal DNA vectors exclusively comprising therapeutically relevant sequences hold great promise for the development of novel therapeutic regimen. Dumbbell-shaped vectors represent non-viral non-integrating DNA minimal vectors which have entered an advanced stage of clinical development (Hardee et al., 2017). Spliceable introns and DNA nuclear import signals such as SV40 enhancer sequences are molecular features that have found multiple applications in plasmid vectors to improve transgene expression. In dumbbells however, effects triggered by introns were not investigated and DNA-based nuclear import sequences have not found applications yet, presumably because dumbbell vectors have continuously been minimized with regard to size. We investigated the effects of an intron and/or SV40 enhancer derived sequences on dumbbell vector driven reporter gene expression. The implementation of a spliceable intron was found to enhance gene expression unconditionally in all investigated cell lines. Conversely, the use of the SV40 enhancer improved gene expression in a cell type-dependent manner. Though both features significantly enlarge dumbbell vector size, neither the intron nor the enhancer or a combination of both revealed a negative effect on gene expression. On the contrary, both features together improved dumbbell-driven gene expression up to 160- or 56-fold compared with plasmids or control dumbbells. Thus, it is highly recommended to consider an intron and the SV40 enhancer for dumbbell vector design. Such an advanced design can facilitate pre-clinical and clinical applications of dumbbell-shaped DNA vectors.
Keywords: Dumbbell vectorBackground
Although many genes have been expressed using dumbbell-shaped DNA vectors, most of these applications used the basic design comprising a promoter, the coding sequence (CDS), and a transcriptional terminator. Some vectors included a chimeric intron, however, it was not reported whether transgene expression was enhanced by this design (Schirmbeck et al., 2001). Here we studied the effects triggered by molecular features that frequently find applications in plasmid design on dumbbell-driven gene expression: 1. A chimeric intron derived from the human β-globin gene–splicing is known to facilitate RNA processing, nuclear export, and subsequently gene expression (Luo and Reed, 1999); and 2. The Simian virus 40 (SV40) enhancer which can enhance the activity of the homologous SV40 promoter and which in addition was reported to function as an active DNA nuclear import sequence (Dean, 1997; Dean et al., 1999). The proposed mechanism behind this phenomenon is that the SV40 enhancer recruits transcription factors harboring protein nuclear import signals in the cytoplasm and that the vector DNA is piggyback translocated into the nucleus with support of the protein nuclear import machinery. We generated luciferase-expressing dumbbell vectors harboring either both, only one or none of these molecular features and monitored transgene expression in HEK293T and HepG2 cells. Our data demonstrate that introns and the SV40 enhancer can substantially improve dumbbell vector design (Jiang et al., 2016).
Materials and Reagents
- Pipette tips (Corning® Isotip® filtered 0.2-10 μl) (Corning, catalog number: 4807 )
- Pipette tips (Axygen® TF200RS 1-200 μl) (Corning, Axygen®, catalog number: TF-200-R-S )
- Pipette tips (Axygen® TF1000RS 100-1,000 μl) (Corning, Axygen®, catalog number: TF-1000-R-S )
- 1.5 ml microcentrifuge tubes (RNase, DNase and Pyrogen-Free) (Corning, Axygen®, catalog number: MCT-150-C )
- 0.2 ml thin-walled PCR tubes (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 3412 )
- FalconTM 50 ml conical centrifuge tubes (Corning, Falcon®, catalog number: 352070 )
- T-75 flask (Corning, catalog number: 3290 )
- 24-well cell culture plate (Corning, catalog number: 3527 )
- 96-well plate (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 442404 )
- HEK293T cells (ATCC, catalog number: CRL-3216 )
- HepG2 cells (ATCC, catalog number: HB-8065 )
- pGL3-control vector (Promega, catalog number: E1741 )
- One Shot® TOP10 Chemically Competent E. coli (Thermo Fisher Scientific, InvitrogenTM, catalog number: C404010 )
- Chimeric human β-globin intron sequence (Gene synthesis, GeneArt, Applied Biosystems): 5’-CAGGTAAGTATCAAGGTTACAAGACAGGTTTAAGGAGACCAATAGAAACTGGGCTTGTCGAGACAGAGACGACTCTTGCGTTTCTGATAGGCACCTATTGGTCTTACTGACATCCACTTTGCCTTTCTCTCCACAGG-3’
- SV40 enhancer sequence: 5’-CGATGGAGCGGAGAATGGGCGGAACTGGGCGGAGTTAGGGGCGGGATGGGCGGAGTTAGGGGCGGGACTATGGTTGCTGACTAATTGAGATGCATGCTTTGCATACTTCTGCCTGCTGGGGAGCCTGGGGACTTTCCACACCTGGTTGCTGACTAATTGAGATGCATGCTTTGCATACTTCTGCCTGCTGGGGAGCCTGGGGACTTTCCACACCCTAACTGACACACATTCCACAGC-3’
- Oligonucleotide primers for chimeric intron amplification (Synthesized by Integrated DNA Technologies, 25 nmol scale, deprotected desalted):
intron-Fw: 5’-ATCTATCGGGATCCAAGCTTCAGGTAAGTATCAAGGTTACAAGACAGG-3’
intron-Rv: 5’-CATTATCTGGATCCCCATGGACCCTGTGGAGAGAAAGGCAA-3’ - Loop sequences (Synthesized by Integrated DNA Technologies, 25 nmol scale, deprotected desalted):
Loop-1: 5’-pGATCTGACCAGTTTTCTGGTCA-3’
Loop-2: 5’-pTCGACAGGCTCTTTTGAGCCTG-3’ - Oligonucleotide primers for poly(A) signal (Synthesized by Integrated DNA Technologies, 25 nmol scale, deprotected desalted):
polyA-Fw: 5’-TGTAATTCTAGAGTCGGGGCG-3’
polyA-Rv: 5’-ATCTATCGGGATCCTTACCACATTTGTAGAGGTT-3’ - FastDigest HindIII (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: FD0504 )
- FastDigest NcoI (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: FD0573 )
- FastDigest BamHI (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: FD0054 )
- FastDigest BglII (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: FD0083 )
- FastDigest XhoI (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: FD0694 )
- FastDigest SalI (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: FD0644 )
- FastDigest AseI (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: FD0914 )
- UltraPureTM DNase/RNase-free distilled water (Thermo Fisher Scientific, InvitrogenTM, catalog number: 10977015 )
- T7 DNA polymerase (10 U/µl) (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: EP0081 )
- Taq DNA polymerase, recombinant (5 U/µl) (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: EP0402 )
- 10x FD buffer (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: B64 )
- T4 DNA ligase (5 U/µl) (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: EL0014 )
- dNTP set 100 mM solutions (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: R0181 )
- QIAquick PCR purification kit (QIAGEN, catalog number: 28106 )
- Potassium acetate (Sigma-Aldrich, catalog number: P1190-100G )
- Agarose, LE, analytical grade (Promega, catalog number: V3125 )
- Ethanol, absolute (Fisher Scientific, catalog number: BP28184 )
- Sodium chloride (NaCl) (Sigma-Aldrich, catalog number: S9888-500G )
- Magnesium chloride hexachloride (MgCl2·6H2O) (Sigma-Aldrich, catalog number: M2670-100G )
- Tris-HCl (Roche Diagnostics, catalog number: 10812846001 )
- Ethylenediaminetetraacetic acid (EDTA) (Sigma-Aldrich, catalog number: EDS-100G )
- Adenosine 5’-triphosphate disodium salt hydrate (Sigma-Aldrich, catalog number: A2383-1G )
- Ethidium bromide solution (Bio-Rad Laboratories, catalog number: 1610433 )
- Phenol solution (Sigma-Aldrich, catalog number: P4557-100ML )
- Chloroform (Sigma-Aldrich, catalog number: 288306-1L )
- 3-Methyl-1-butanol (Sigma-Aldrich, catalog number: 309435-100ML )
- GeneRuler DNA Ladder Mix (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: SM0331 )
- HyClone Dulbecco’s modified Eagles medium/high glucose with L-glutamine, sodium pyruvate (GE Healthcare, HyCloneTM, catalog number: SH30243.01 )
- HyClone Standard Fetal Bovine Serum (GE Healthcare, HyCloneTM, catalog number: SH30088.03 )
- Penicillin-streptomycin (10,000 U/ml) (Thermo Fisher Scientific, GibcoTM, catalog number: 15140122 )
- Lipofectamine® 2000 Transfection Reagent (Thermo Fisher Scientific, InvitrogenTM, catalog number: 11668019 )
- Opti-MEM® I Reduced Serum Medium (Thermo Fisher Scientific, GibcoTM, catalog number: 31985070 )
- Luciferase Assay System (Promega, catalog number: E1501 )
- Liquid nitrogen
- TE buffer (see Recipes)
Equipment
- Pipettes (Gilson, PIPETMAN Classic®, P2 , P20N , P200N , and P1000N )
- CO2 incubator (Thermo Electron)
- Shaker (Heidolph Instruments, model: Unimax 2010 )
- Glass beaker (Schott, Duran)
- Standard thermal cycler (Thermo Fisher Scientific, Applied BiosystemsTM, model: GeneAmp PCR System 9700 )
Note: This product has been discontinued. - Gel doc (Bio-Rad Laboratories, Gel Doc Imager)
- Gel running apparatus (Amersham Biosciences)
- Gel staining tray (GE Healthcare)
- Benchtop centrifuge (Eppendorf, model: 5430 R )
- Heat block (Thermomixer comfort) (Eppendorf)
- Spectrophotometer (Thermo Fisher Scientific, Thermo ScientificTM, model: NanoDropTM 2000 )
- Microwave (Panasonic)
- Class II Biological Safety Cabinet (Gelman)
- Synergy H1 Multi-Mode Reader (BioTek Instruments, model: Synergy H1 )
Software
- GraphPad Prism software 5.0
Procedure
- Design and molecular cloning of luciferase-expressing plasmid variants
To study the function of a synthetic intron and/or the SV40 enhancer on dumbbell-driven gene expression, we clone the corresponding plasmid vectors based on the pGL3-control vector. The pGL3-control vector harbors the firefly luciferase gene under the control of the SV40 promoter together with the SV40 enhancer element at the 3’ end of the expression cassette. Cloning is done as described below:- The 137 bp chimeric human β-globin mini-intron is synthesized by gene synthesis (GeneArt, Applied Biosystems), PCR amplified using primers intron-Fw and intron-Rv, and then inserted into the pGL3-control plasmid using the HindIII and NcoI sites to create a luciferase expression vector featured with both, an intron and the SV40 enhancer (int-luc-enh) (Jiang et al., 2016 and 2017). An illustration of the production process is shown in Figure 1.

Figure 1. Illustration of the plasmid cloning schemes. Step 1: Plasmid p-int-luc-enh is derived by insertion of the b-globin mini-intron into the HindIII and NcoI sites of vector pGL3-Control (p-luc-enh). Step 2: Plasmid p-luc can be generated by exchanging the SV40 enhancer together with the SV40 poly(A) site of vector pGL3-Control (p-luc-enh) by the SV40 polyA site using XbaI and BamHI. Step 3: Plasmid p-int-luc is cloned by exchanging the SV40 enhancer together with the SV40 polyA site of vector p-int-luc-enh by the SV40 polyA site using XbaI and BamHI. - The SV40 enhancer sequence in pGL3-control is deleted by digestion of the vector with XbaI and BamHI. By doing so, we also delete the SV40 late poly(A) signal. To retrieve the SV40 poly(A) signal, its sequence is PCR-amplified using the primers polyA-Fw and polyA-Rv which introduce the XbaI and BamHI sites. The PCR product is cleaved with XbaI and BamHI and the SV40 poly (A) signal is re-inserted to create a plasmid lacking both, the intron and the SV40 enhancer (luc) (Jiang et al., 2016 and 2017). An illustration of the production process is shown in Figure 1.
- The equivalent procedure described under step A2 is repeated starting with the pGL3-control variant harboring the intron and the SV40 enhancer to generate a plasmid that is only featured with the intron (int-luc) (Jiang et al., 2016). An illustration of the production process is shown in Figure 1.
- For simplification, novel names are assigned to the constructs as listed in Table 1.
Table 1. Nomenclature of luciferase expression constructs. Plasmids were assigned the prefix ‘p’, dumbbells the prefix ‘db’.
- The 137 bp chimeric human β-globin mini-intron is synthesized by gene synthesis (GeneArt, Applied Biosystems), PCR amplified using primers intron-Fw and intron-Rv, and then inserted into the pGL3-control plasmid using the HindIII and NcoI sites to create a luciferase expression vector featured with both, an intron and the SV40 enhancer (int-luc-enh) (Jiang et al., 2016 and 2017). An illustration of the production process is shown in Figure 1.
- Production of luciferase-expressing dumbbell vectors
- Luciferase-expressing dumbbell vectors are produced from the corresponding plasmids described under Procedure A. using the enzymatic ligation assisted by nucleases (ELAN) loop-ligation method (Cost, 2007). In brief, the gene expression cassette is directly cut out from the respective parental plasmid. 5’ phosphorylated loop-forming oligonucleotides designed following the protocol by Cost are then ligated to form the dumbbell-shaped structure. An illustration of the production process is shown in Figure 2.
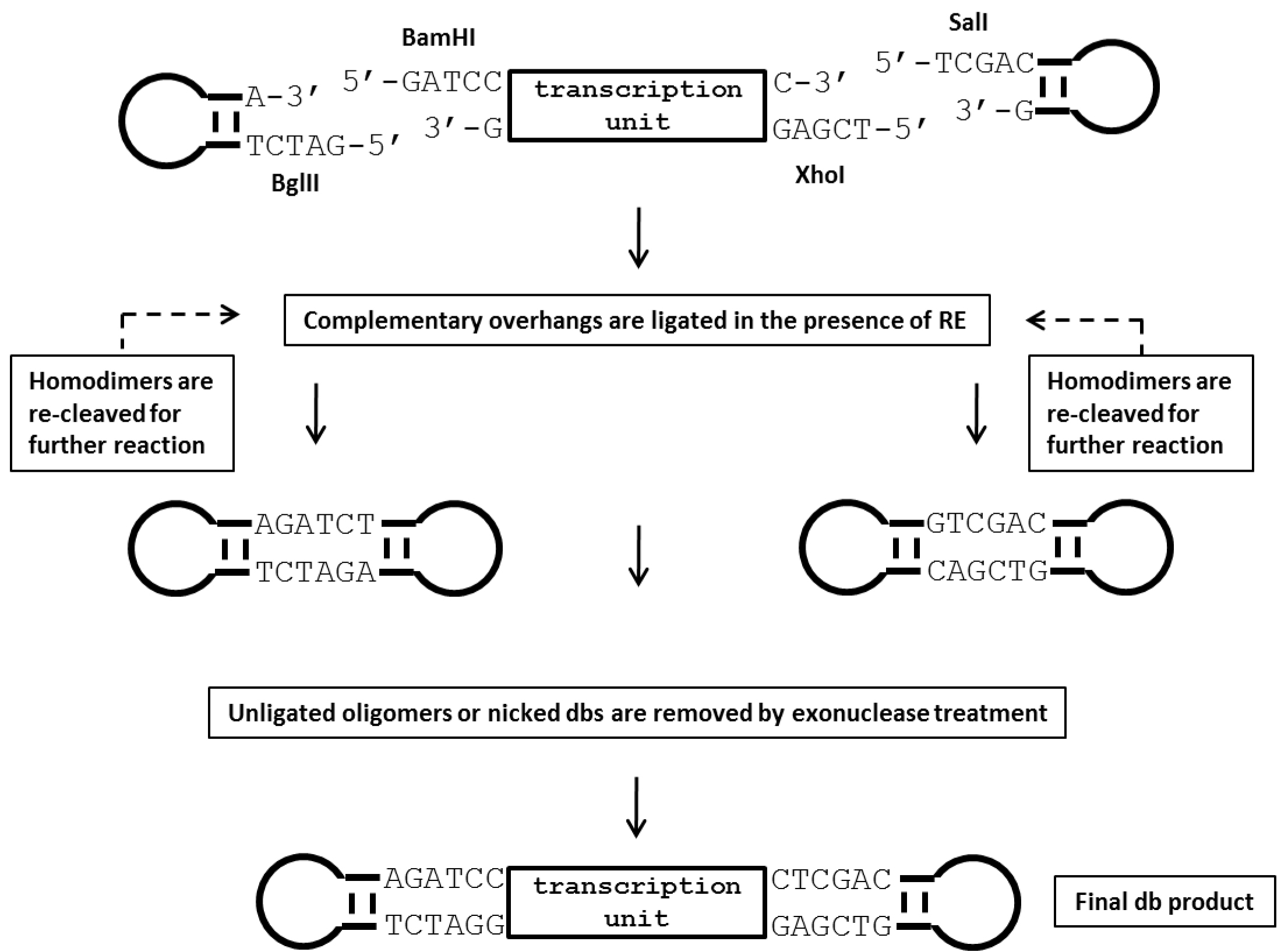
Figure 2. Illustration of the production process for dumbbell vectors using the ELAN loop-ligation method. In this strategy, a parental plasmid containing the transgene expression cassette is first digested with two restriction endonucleases (BamHI and XhoI in this example) that recognize sites flanking the transcription unit. Next, 5’ phosphorylated oligonucleotides (ODNs) forming loop-structures with compatible restriction overhangs are ligated in the presence of the four restriction enzymes BglII, BamHI, XhoI and SalI which cleave homodimers formed by the loop ODN or the transcription unit. This strategy assists the generation of the correctly ligated dumbbell vector. Non-ligated sequences are subsequently destroyed by T7 DNA polymerase treatment, which exhibits strong 3’-5’ exonuclease activity (Engler and Richardson, 1983). RE, restriction enzyme. - For the ELAN ligation reaction, loop ODN is added in a 50-fold molar excess over the linear expression unit DNA. Misligated by-products such as dimers formed of the loops or the transcription unit are cleaved by a set of four restriction enzymes (BamHI, BglII, XhoI, SalI) to foster dumbbell formation. A fifth restriction enzyme (AseI) is added to cleave dumbbells formed by ligation of the loop ODN with the plasmid backbone. This fifth enzyme can be a single or ideally a multi-cutter with respect to the cloning vector backbone but must not cut within the dumbbell vector. Non-ligated sequences are removed by exonuclease treatment. Detailed setups of the reactions are summarized in Table 2.
Table 2. Three-step reaction procedure for the generation of luciferase-expressing dumbbells using the ELAN loop-ligation strategy. Step 1: Excision of the expression cassette from the respective parental plasmid, in this example using the enzymes XhoI and BamHI. Step 2: ELAN reaction including the T4 DNA ligase and five restriction endonuclease, in this example BglII, SalI, BamHI, XhoI, and AseI. Step 3: Exonuclease reaction using the T7 DNA polymerase. It is recommended to add again the restriction endonucleases of step 2 to ensure that any dumbbell-shaped byproducts taken over from step 2 are cleaved and rendered amendable for exonuclease degradation. After steps 1 and 2, enzymes are heat-inactivated but no further purification is needed.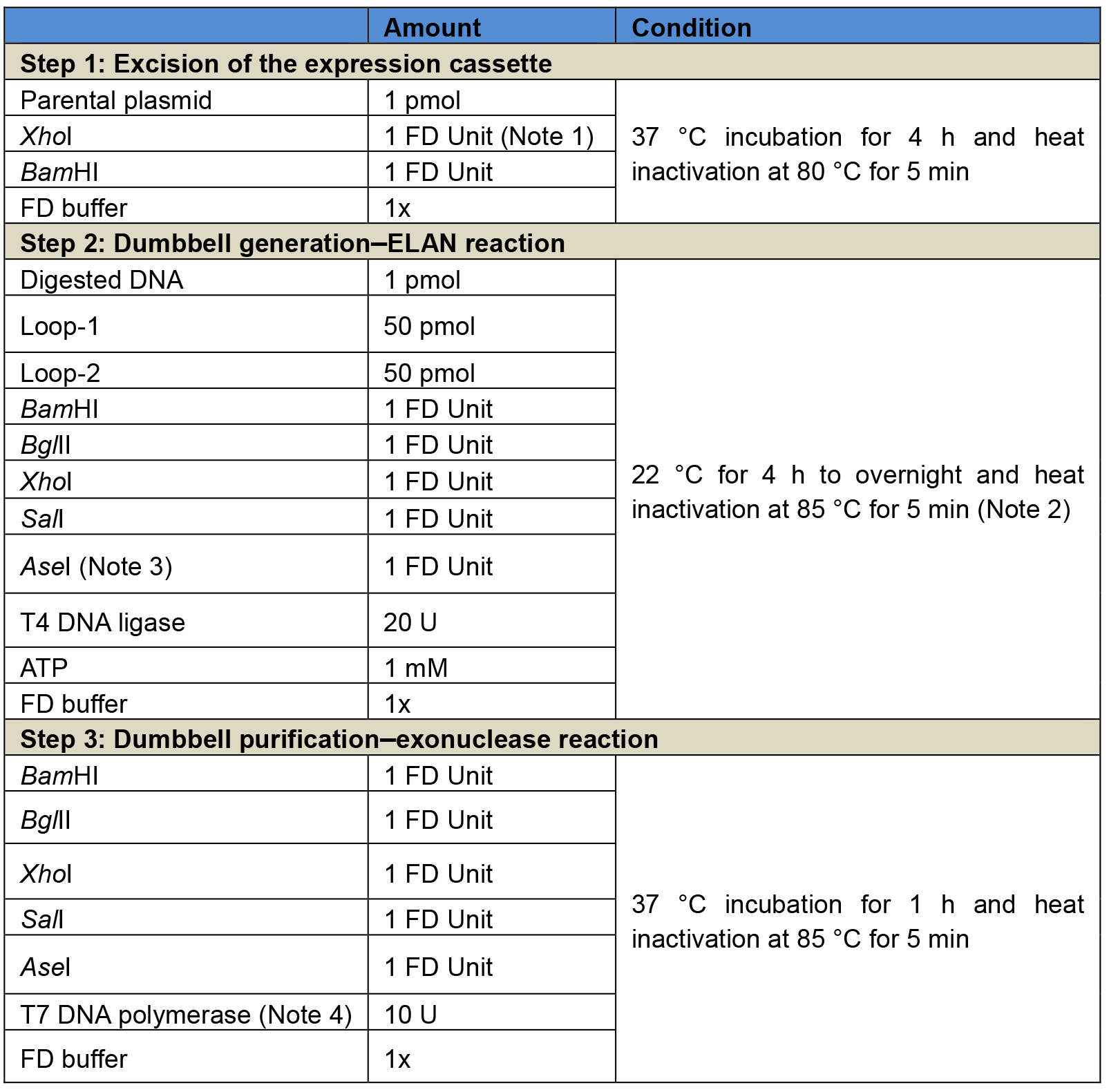
- Quantity and quality of dumbbell vectors is analyzed by agarose gel electrophoresis as shown in Figure 3. Prior to the transfection of cells, dumbbell DNA is purified using the QIAquick PCR purification kit (QIAGEN) using the manufacturer’s instructions followed by ethanol precipitation. For the ethanol precipitation, the elution volume of 50 µl is topped up with distilled water to 400 µl. Then 40 µl of 3 M potassium acetate (pH 5.0) and 2.5 volumes (1,100 µl) of absolute ethanol are added, the solution is mixed by inverting the tube and placed for precipitation at -20 °C for 20 min. Vector DNA is pelleted by centrifugation at 16,100 x g for 15 min. The pellets are washed with 500 µl 4 °C 70% ethanol and air-dried. Purified DNA is then dissolved in TE buffer or distilled water.
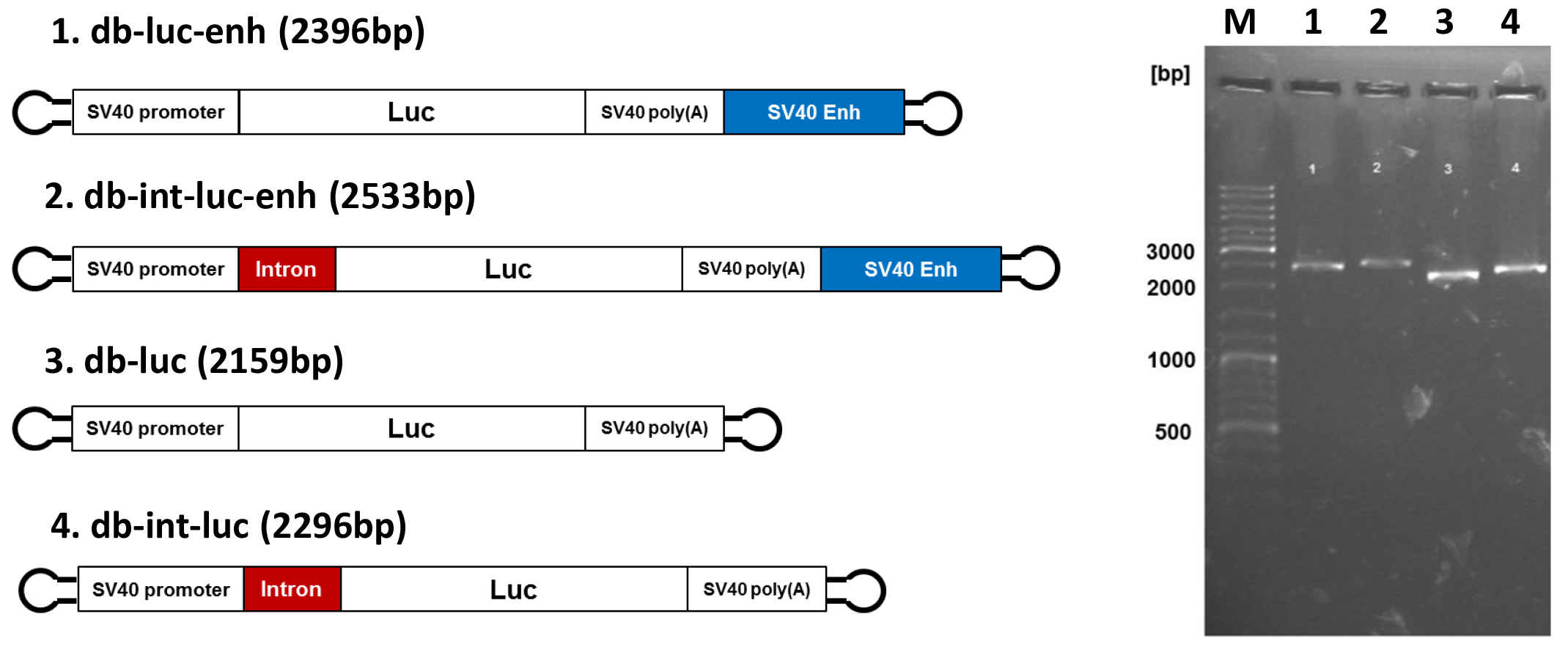
Figure 3. Dumbbell vectors for enhanced gene expression. Left side. Design of advanced dumbbell vectors: db-luc: basic vector; db-luc-enh: dumbbell featured with the SV40 enhancer; db-int-luc: dumbbell featured with an intron; db-int-luc-enh: dumbbell featured with SV40 enhancer and intron. Right side. Analytical 0.8% agarose gel electrophoresis of the four luciferase dbs after exonuclease treatment. The expected dumbbell bands but no by-products are detected. Numbers of lanes on the right correspond to the dumbbell numbers on the left. M: GeneRuler DNA Ladder Mix (Thermo Fisher Scientific).
- Luciferase-expressing dumbbell vectors are produced from the corresponding plasmids described under Procedure A. using the enzymatic ligation assisted by nucleases (ELAN) loop-ligation method (Cost, 2007). In brief, the gene expression cassette is directly cut out from the respective parental plasmid. 5’ phosphorylated loop-forming oligonucleotides designed following the protocol by Cost are then ligated to form the dumbbell-shaped structure. An illustration of the production process is shown in Figure 2.
- Transfection of human tissue culture cells with luciferase-expressing plasmids and dumbbells
- HEK293T and HepG2 cells are cultured in Dulbecco’s modified Eagle’s medium (DMEM, Invitrogen) supplemented with 10% (v/v) heat-inactivated fetal bovine serum (Hyclone) and 1% penicillin-streptomycin solution (Invitrogen). Cells are kept in humidified incubator with 5% CO2, and are passaged at 80-90% confluency.
- Cells are transfected using Lipofectamine 2000 (Invitrogen) following the manufacturer’s protocol. In brief, cells are seeded in 24-well plates one day prior to transfection at 70-80% confluency. At the day of transfection, 400 ng DNA is mixed with 1 µl Lipofectamine 2000 in Opti-MEM (Invitrogen) using a total volume of 100 µl. The DNA-liposome complexes are formed at room temperature during a 20 min incubation time, and then the mixture is added dropwise to the cells.
- Transfected cells are harvested and analyzed 48 h post transfection.
- HEK293T and HepG2 cells are cultured in Dulbecco’s modified Eagle’s medium (DMEM, Invitrogen) supplemented with 10% (v/v) heat-inactivated fetal bovine serum (Hyclone) and 1% penicillin-streptomycin solution (Invitrogen). Cells are kept in humidified incubator with 5% CO2, and are passaged at 80-90% confluency.
- Luciferase assays
Measuring firefly luciferase enzyme activity in cell lysates using the luciferase assay system from Promega:- For cell lysis, tissue culture medium is removed and 100 µl of passive lysis buffer (PLB, provided by the manufacturer) is added to each 24-well. Cells are incubated for 20 min at room temperature applying gentle shaking.
- After the lysis procedure, cell lysates are aliquoted, immediately frozen in liquid nitrogen, and stored at -80 °C. Though not further specified by the provider, frozen samples are stable for months or years.
- For the luciferase assay, 10 µl of each sample is transferred into an opaque white 96-well plate (Nunc).
- The Synergy H1 Hybrid Multi-Mode Microplate Reader (BioTek) system is used to inject Luciferase Assay Reagent (LARII) automatically and read the luminescence. Background signals are determined by blank and water controls for each assay and luciferase activity is normalized according to recommendations from Promega (Schagat et al., 2007).
- Representative data are shown in Figure 4.
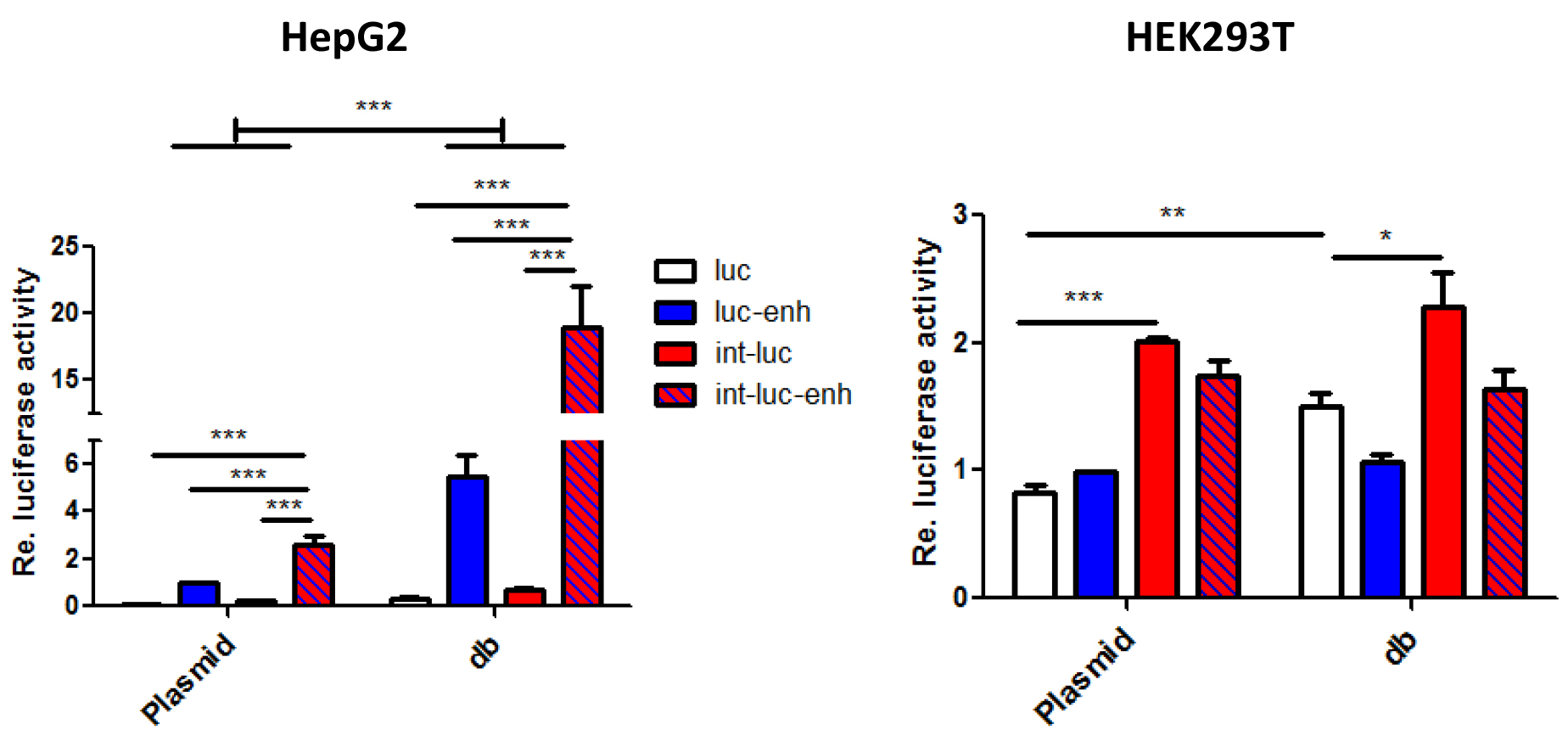
Figure 4. Enhancement of dumbbell vs. plasmid-driven luciferase expression by the β-globin gene chimeric intron and/or the full length SV40 enhancer. 5 x 104 HEK293T or HepG2 cells seeded in 24-wells were transfected with 400 ng dumbbell vectors or equivalent amounts of parental plasmids. Luciferase expression triggered by equimass amounts of dumbbell vectors and plasmids measured 48 h post transfection. Error bars indicate mean deviations from average of three to five independent experiments. Re: Relative.
- For cell lysis, tissue culture medium is removed and 100 µl of passive lysis buffer (PLB, provided by the manufacturer) is added to each 24-well. Cells are incubated for 20 min at room temperature applying gentle shaking.
Data analysis
Prism 5.0 GraphPad software is used for the data presentation and statistical analysis. Luciferase assay results are shown as mean ± SEM. For the comparison of the data, one-way ANOVA with Newman-Keuls post hoc test is used. * represents P value < 0.05, ** represents P value < 0.01, and *** represents P value < 0.001.
Notes
- 1 FD Unit is defined as 1 µl FastDigest enzyme by Thermo Fisher Scientific.
- According to our experience, the ligation reaction is completed within 4 h and longer incubation times do not improve the dumbbell yield.
- AseI is chosen to destroy the bacterial pGL3-Control plasmid backbone because it does not cleave within the dumbbell vector sequences comprising the gene of interest, the SV40 enhancer, and the intron. The choice of this enzyme however depends on the respective sequences of the dumbbell and the cloning vector backbone.
- T7 DNA polymerase exhibits 100% activity in the FD buffer and is therefore directly added into the ligation mixture.
Recipes
- TE buffer
10 mM Tris-HCl, pH 8.0
1 mM EDTA
Acknowledgments
The protocol described herein was developed and utilized previously in Jiang et al. (2016). This work was supported by the National University of Singapore [Bridging Grant NUHSRO/2015/091/Bridging/02], the National Medical Research Council of Singapore [New Investigator Grant number NMRC/NIG/1058/2011], and the Ministry of Education of Singapore [Academic Research Fund (AcRF) Tier 1 Faculty Research Committee (FRC) grants number T1-2011Sep-04 and T1-2014Apr-02 and Seed Fund for Basic Science Research number T1-BSRG 2015-05], all to VP. The authors declare competing financial interests. A patent application covering major parts of the work is pending.
References
- Cost, G. J. (2007). Enzymatic ligation assisted by nucleases: simultaneous ligation and digestion promote the ordered assembly of DNA. Nat Protoc 2(9): 2198-2202.
- Dean, D. A. (1997). Import of plasmid DNA into the nucleus is sequence specific. Exp Cell Res 230(2): 293-302.
- Dean, D. A., Dean, B. S., Muller, S. and Smith, L. C. (1999). Sequence requirements for plasmid nuclear import. Exp Cell Res 253(2): 713-722.
- Engler, M. J. and Richardson, C. C. (1983). Bacteriophage T7 DNA replication. Synthesis of lagging strands in a reconstituted system using purified proteins. J Biol Chem 258(18): 11197-11205.
- Hardee, C. L., Arevalo-Soliz, L. M., Hornstein, B. D. and Zechiedrich, L. (2017). Advances in non-viral DNA vectors for gene therapy. Genes (Basel) 8(2).
- Jiang, X. and Patzel, V. (2017). Formation of minimized hairpin template-transcribing dumbbell vectors for small RNA expression. Bio Protoc 7(11): e2313.
- Jiang, X., Yu, H., Teo, C. R., Tan, G. S., Goh, S. C., Patel, P., Chua, Y. K., Hameed, N. B., Bertoletti, A. and Patzel, V. (2016). Advanced design of dumbbell-shaped genetic minimal vectors improves non-coding and coding RNA expression. Mol Ther 24(9): 1581-1591.
- Luo, M and Reed, R (1999). Splicing is required for rapid and efficient mRNA export in metazoans. Proc Natl Acad Sci USA 96: 14937-14942.
- Schagat, T., Paguio, A. and Kopish, K. (2007). Normalizing genetic reporter assays: approaches and considerations for increasing consistency and statistical significance. Cell Notes 9-12.
- Schirmbeck, R., Konig-Merediz, S. A., Riedl, P., Kwissa, M., Sack, F., Schroff, M., Junghans, C., Reimann, J. and Wittig, B. (2001). Priming of immune responses to hepatitis B surface antigen with minimal DNA expression constructs modified with a nuclear localization signal peptide. J Mol Med (Berl) 79(5-6): 343-350.
Article Information
Copyright
© 2017 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Jiang, X. and Patzel, V. (2017). Advanced Design of Minimalistic Dumbbell-shaped Gene Expression Vectors. Bio-protocol 7(15): e2425. DOI: 10.21769/BioProtoc.2425.
Category
Microbiology > Microbial biochemistry > DNA
Molecular Biology > DNA > Gene expression
Molecular Biology > DNA > DNA cloning
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.