- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Endpoint or Kinetic Measurement of Hydrogen Sulfide Production Capacity in Tissue Extracts
Published: Vol 7, Iss 13, Jul 5, 2017 DOI: 10.21769/BioProtoc.2382 Views: 11498
Reviewed by: Neelanjan BoseTanxi CaiAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Jan 2015

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
Hydrogen sulfide (H2S) gas is produced in cells and tissues via various enzymatic processes. H2S is an important signaling molecule in numerous biological processes, and deficiencies in endogenous H2S production are linked to cardiovascular and other health complications. Quantitation of steady-state H2S levels is challenging due to volatility of the gas and the need for specialized equipment. However, the capacity of an organ or tissue extract to produce H2S under optimized reaction conditions can be measured by a number of current assays that vary in sensitivity, specificity and throughput capacity. We developed a rapid, inexpensive, specific and relatively high-throughput method for quantitative detection of H2S production capacity from biological tissues. H2S released into the head space above a biological sample reacts with lead acetate to form lead sulfide, which is measured on a continuous basis using a plate reader or as an endpoint assay.
Keywords: Hydrogen sulfide production capacityBackground
Hydrogen sulfide (H2S) gas is produced endogenously by at least three different enzymes in mammals (CGL, CBS, 3-MST) with a range of tissue and cell-type distributions. H2S functions as a gasotransmitter and effector molecule (Wang, 2012) in a wide range of biological functions related to metabolism (Módis et al., 2013), stress resistance (Hine et al., 2015), and redox biology (Dickhout et al., 2012). Reduced H2S is linked to cardiovascular problems including hypertension in rodents (Yang et al., 2008) and cardiac hypertrophy in man (Polhemus et al., 2014). Increased H2S can also cause pathology, for example in rodent pancreatitis (Bhatia et al., 2005). Thus, accurate and quantitative detection of H2S from biological sources could facilitate a better understanding of its biological effects as well as its potential use as a clinical biomarker.
Techniques to measure absolute concentrations of H2S present in biological samples, along with their pros and cons, have been reviewed extensively (Olson, 2012; Wang, 2012; Hartle and Pluth, 2016; Takano et al., 2016). For example, free and sulfane-bound H2S pools can be measured in biological samples including serum or tissue homogenates ex vivo using headspace GC-MS, which is highly sensitive and selective, but requires expensive equipment. Nonetheless, due to the volatility of H2S, its interaction with other biological macromolecules and its breakdown into different sulfur-containing compounds, quantitative detection of steady-state free H2S levels in vivo remains challenging (Olson, 2009).
An alternate approach is to measure the capacity of a tissue homogenate or extract to produce H2S in a reaction mixture containing optimized levels of substrate and cofactor, thus allowing for H2S detection methods that are specific but less sensitive. An example is the methylene blue method in which H2S in solution is trapped by lead acetate to form lead sulfide, which upon conversion to methylene blue can be easily read in a standard spectrophotometer (Stipanuk and Beck, 1982; Ikeda et al., 2017). The pros and cons that must be taken into account with each method are based on the question being asked, the biological system and tissue being studied, the relative need for sensitivity, selectivity, or speed, and the cost and resources of the investigator.
Here, we describe an inexpensive, rapid, and moderately high throughput methodology for measuring H2S production capacity in extracts of relatively small amounts of biological material. This method is based on the reaction of H2S present in the headspace above a biological sample with lead acetate to form the black precipitate lead sulfide, a technique used throughout the past 100 years to detect H2S and H2S-producing bacteria (McBride and Edwards, 1914; Kuester and Williams, 1964; Zhang and Weiner, 2014). Previously, we used this method to detect changes in H2S production capacity as a function of diet or genetic background in a variety of biological samples including yeast, worms, flies, and rodent tissues/organs including liver (Hine et al., 2015; Mitchell et al., 2016; Nikonorova et al., 2017). Here, we present an optimized procedure to measure H2S production capacity in mammalian liver via (B) an end-point assay using Whatman paper-embedded lead acetate, or (C) a kinetic assay using agar-embedded lead acetate. As the liver is a strong producer of H2S in mammalian systems via the enzyme cystathionine gamma lyase (CGL) (Kabil et al., 2011), we feel this is a good starting point for researchers to understand and confidently develop this protocol for their own research questions. Furthermore, this procedure can be easily adapted to other biological samples and organisms, although the procedure may need to be optimized by the investigator in order to obtain suitable results.
Materials and Reagents
- Petri dish
- 1.5 ml RNase-free disposable pellet pestles and 1.5 ml tubes (Fisher Scientific, catalog number: 12-141-368 )
- Disposable razor blades
- 8-strip well format tubes (Denville Scientific)
- Hard/rigid plastic dissecting platform/sheet
- Plastic wrap
- Filter paper (703 Style Whatman)
- 96-well plates with lid (Corning, catalog number: 3370 )
- Gloves and proper personal protective equipment
- 15 ml centrifuge tube
- Flash frozen mouse livers
- De-ionized water
- Phosphate buffered saline (PBS), pH 7.4 (Fisher Scientific, catalog number: BP24384 )
- Liquid nitrogen
- Ice
- Dry ice
- 5x passive lysis buffer (Promega, catalog number: E1941 )
- BCA Protein Assay Kit (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 23227 )
- L-cysteine (Sigma-Aldrich, catalog number: C7352 )
- Pyridoxal 5’-phosphate hydrate (Sigma-Aldrich, catalog number: P9255 )
- Lead(II) acetate trihydrate (Sigma-Aldrich, catalog number: 316512 )
- Agarose (HS Molecular Biology Grade) (Denville Scientific, catalog number: CA3510-8 , or use similar)
Note: This product has been discontinued. - 1x passive lysis buffer (see Recipes)
- 20 mM lead(II) acetate trihydrate (see Recipes)
- H2S reaction mixture (see Recipes)
- 1% agarose gel with 100 mM lead(II) acetate trihydrate (see Recipes)
Equipment
- Liquid nitrogen flask (Thermo flask 2122) (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 2122 )
- Forceps
- Scale (OHAUS, catalog number: EP214C )
- Pipettes and tips (single use and multichannel for pipetting between 2 µl to 5 ml)
- -80 °C freezer
- Motorized tissue grinder (Fisher Scientific, catalog number: 12-1413-61 )
- 37 °C water bath
- Micro-centrifuge (VWR, model: Galaxy 16DH )
- UV-Vis plate reader (BioTek Instruments, model: Synergy 2 )
- Large glass Pyrex baking dish (> 100 ml)
- Glass flask (> 100 ml capacity)
- Vacuum oven (VWR, catalog number: 89508-424 )
- Incubator (VWR, model: 1500E )
- Digital camera (Kodak, model: KODAK EASYSHARE C182 )
- Vortex mixer (Scientific Industries, model: Vortex-Genie 2 , catalog number: SI-0236)
- Computers (HP Pavilion dv6 and Lenovo IdeaPad)
- Heat block cube (9.5 x 7.5 x 5 cm), or other heavy object with approximate dimensions
Software
- GraphPad Prism 7
- Microsoft Excel
- ImageJ
- Gen5
Procedure
Note: This protocol includes two related techniques to measure H2S production capacity via an endpoint assay (Procedure B) or a kinetic assay (Procedure C). Both techniques use the specific chemistry in which lead acetate (white in color) is converted to lead sulfide (brown to black in color) upon exposure the H2S. The techniques differ in their detection methods, with (Procedure B) utilizing endpoint densitometry to quantitate lead sulfide spots on the filter paper, while (Procedure C) utilizes the kinetic-capable absorbance of light in the 310 nm wavelength as well as endpoint densitometry of the lead sulfide spots on the agar gel. Shared common procedures and techniques are presented first (Procedure A) and then later split into the specifics that differentiate the two.
- Shared procedures
- Tissue samples
- Excise mouse liver tissue following euthanasia according to the procedures detailed in the animal use protocol that has been approved by your institutional animal welfare committee.
- Before euthanasia and harvesting, prepare 10 ml PBS in a Petri dish on ice to use as a wash for removing superficial blood/debris from the excised liver tissues. Also, fill the liquid nitrogen flask with liquid nitrogen.
- After euthanasia, quickly remove the large left lobe of the liver, rinse in PBS, place into a 1.5 ml centrifuge tube and flash freeze in liquid nitrogen.
- Leave samples in liquid nitrogen until all livers have been harvested.
- Before euthanasia and harvesting, prepare 10 ml PBS in a Petri dish on ice to use as a wash for removing superficial blood/debris from the excised liver tissues. Also, fill the liquid nitrogen flask with liquid nitrogen.
- For previously harvested liver samples, store at -80 °C until ready for homogenization.
- Excise mouse liver tissue following euthanasia according to the procedures detailed in the animal use protocol that has been approved by your institutional animal welfare committee.
- Homogenize mouse livers
- With liver samples in 1.5 ml centrifuge tubes, remove from liquid nitrogen or -80 °C freezer and place on dry ice.
- Using a sterile or clean disposable razor and forceps, quickly remove the frozen liver from the tube, cut off segments on a hard plastic or other dissecting platform/sheet that is placed on dry ice, and weigh on a scale to approximately 100 mg.
Note: This weight does not need to be accurate, just close enough so that the weight of liver sample to volume of lysis buffer is relatively constant between samples. - While still frozen, place the ~100 mg of liver tissue into a new 1.5 ml centrifuge tube precooled on dry ice.
- Make up a fresh 1x solution of passive lysis buffer (see Recipes) from the 5x passive lysis buffer stock (Promega) with deionized H2O.
Note: 1x passive lysis buffer should be kept on regular ice and made fresh with each assay. The 5x passive lysis buffer should always be stored at -20 °C when not in use. Unfortunately the recipe is proprietary. - One 1.5 ml tube at a time, add 250 µl of 1x passive lysis buffer to each approximate 100 mg sample.
- Immediately, start homogenizing with the Motorized tissue grinder (Fisherbrand) with Pellet Pestle (Fisherbrand) until the sample has been thoroughly homogenized. This usually takes 30 sec to 1 min to perform.
- Remove the grinder and pestle from the tube, secure the cap, and place the homogenized sample and 1.5 ml tube into liquid nitrogen to flash freeze.
- Continue to homogenize all samples, keeping those completed in the liquid nitrogen.
- Once all samples have been homogenized, remove all samples at the same time from liquid nitrogen and thaw at room temperature for approximately 5-10 min or in a 37 °C water bath for approximately 1-5 min.
Caution: Take care to use tubes suitable to freeze/thaw with liquid N2 to avoid loss of samples and potential injury from exploding tubes. - Repeat freeze-thaw an additional two times, ensuring at least 1 min of freezing in the liquid N2 and keeping the same times for thawing as described above.
- Spin down the tubes in a microcentrifuge for 5 min at 4 °C at 5,000 x g.
- Remove and save the supernatants (~200 µl) in 8-strip well format tubes and place on ice if immediately going onto the next step (protein concentration determination). If not, flash freeze the tubes and place into a -80 °C freezer until ready to do protein concentration determination.
- With liver samples in 1.5 ml centrifuge tubes, remove from liquid nitrogen or -80 °C freezer and place on dry ice.
- Protein concentration determination and normalization of protein concentrations
- In fresh 8-trip well tubes, place 198 µl of deionized water and 2 µl of the liver homogenate supernatant to make a 1:100 dilution.
- Follow the directions of the Pierce BCA Protein Assay Kit for preparing the working solutions and standard BSA solutions (between 0 µg/µl to 2 µg/µl) that are contained in the kit for the protein concentration assay.
- Pipette 200 µl of the assay solution in each needed well, and 5 µl of the standard or 1:100 diluted sample.
- Cover with plastic wrap and incubate at room temperature in the dark for 30 min.
- Unwrap the plate and place it onto a UV-Vis spectrophotometer.
Note: We use a Synergy 2 multimode plate reader running the Gen5 software, but other plate readers can be used. - Measure the absorbance of light at 562 nm.
- Determine the protein concentration of each sample based on the standard curve equation obtained from the absorbance at 562 nm from each of the standards.
- In new 8 strip well tubes, normalize the protein concentration of each sample to 20 µg/µl using 1x passive lysis buffer. Bring up the final volume to 100 µl.
Note: If 20 µg/µl is not possible, then dilute each sample to 10 µg/µl or other suitable concentration.
- In fresh 8-trip well tubes, place 198 µl of deionized water and 2 µl of the liver homogenate supernatant to make a 1:100 dilution.
- Tissue samples
- Filter paper-embedded lead acetate endpoint assay
- Prepare lead acetate filter papers
- In a large glass Pyrex dish, pour 100 ml of 20 mM lead(II) acetate trihydrate (see Recipes) in deionized H2O.
- Cut the 703 style Whatman filter papers into approximate size dimensions of a 96-well plate (8 x 12.5 cm).
- Place the cut filter papers into the lead(II) acetate trihydrate solution and let soak for at least 20 min.
- Embed and dry the lead(II) acetate trihydrate into the filter papers by placing the wet papers into the vacuum oven set at 110 °C and the vacuum turned on. Let the papers sit in the vacuum oven at these settings for 20-30 min.
- Store the finished papers at room temperature in a dark and dry area until ready for use. Although we have not empirically tested the shelf-life and stability of these papers, they are good for at least one month after their production.
- In a large glass Pyrex dish, pour 100 ml of 20 mM lead(II) acetate trihydrate (see Recipes) in deionized H2O.
- Prepare H2S production capacity reaction solution.
Note: This must be done fresh for each assay.- Calculate the volume needed to run all of the samples. Each sample requires 150 µl of reaction solution (see Recipes).
- Prepare working stock of L-cysteine (100 mM in PBS) and pyridoxal 5’-phosphate (PLP) (10 mM in PBS). The cysteine must be made fresh each time. For PLP, warming to 37 °C and vortexing may be necessary to completely solubilize the powder. The PLP stock solution can be stored in aliquots at -20 °C for several months.
- In an adequate amount of PBS to cover the needed volume of reaction mixture, add L-cysteine to a final concentration of 10 mM and pyridoxal 5’-phosphate hydrate to a final approximate concentration of 1 mM.
- Keep at room temperature and use immediately.
- Calculate the volume needed to run all of the samples. Each sample requires 150 µl of reaction solution (see Recipes).
- Setting up the assay
- In each well of a 96-well plate, pipette 150 µl of the reaction mixture.
- In each well, pipette 0-500 µg of sample protein. We have found 100 µg (or 5 µl when samples are diluted to 20 µg/µl) produces a detectable and non-saturating signal within 1 h with liver lysates. Other tissues may require more protein.
- Once all samples have been added, ensure there is no residual or spilled liquid on the surface of the 96-well plate by gently wiping with tissue paper.
- Place the dry lead acetate embedded filter paper directly over the 96-well plate.
- Place a similar sized piece of hard/rigid plastic or other non-flexible cover over the paper to keep it in place and ensure direct contact of the lead acetate paper with the lips of the wells on the plate.
- Place the heavy heating block cube or other heavy object with similar dimensions (9.5 x 7.5 x 5 cm) over the piece of hard plastic.
- Making sure not to shake or spill any of the liquid, place the ‘sandwich’ into a non-shaking/rocking 37 °C incubator.
- Over time, brown to black circles will form on the filter paper over the wells (see Figure 1 for visual example). This is the formation of lead sulfide due to the interaction of lead acetate with hydrogen sulfide.
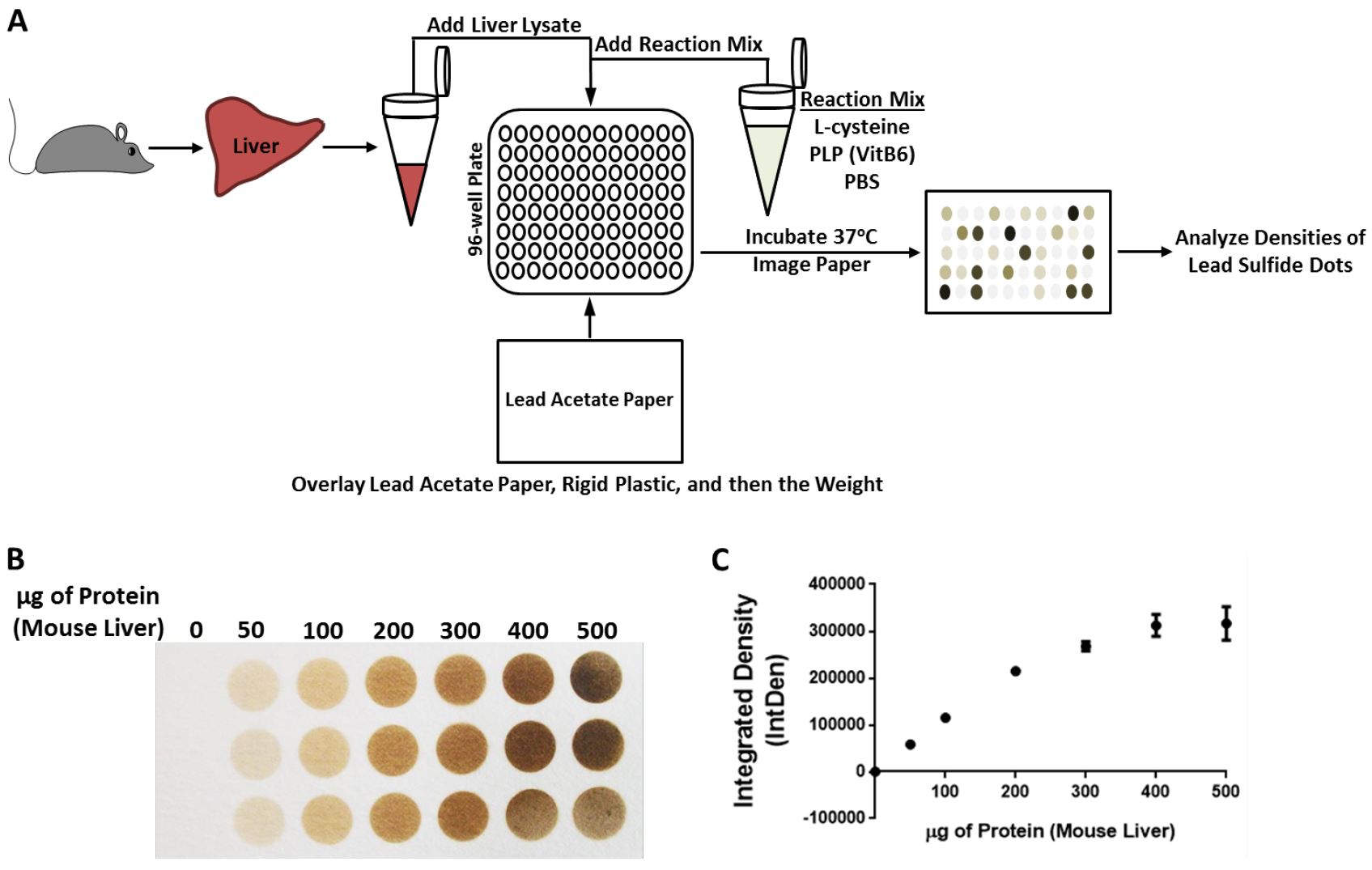
Figure 1. Filter paper-embedded lead acetate endpoint assay for H2S production capacity determination from liver extracts. A. Schematic overview of the assay. Liver is excised from the mouse, homogenized and lysed in passive lysis buffer followed by protein determination and normalization. In a 96-well plate, the reaction mixture is added first, followed by the liver lysate samples. The lead acetate paper, rigid plastic, and weight are then placed on top of the 96-well plate. Incubation occurs at 37 °C for 1 to 2 h followed by imaging and analysis of the lead sulfide dots on the paper. B. Photographic image of the lead acetate embedded filter paper after a 2-h incubation at 37 °C over a 96-well plate containing increasing amounts (between 0 to 500 µg) of mouse liver protein in each in a 150 µl reaction in PBS containing 10 mM L-cysteine and 1 mM pyridoxal 5’-phosphate hydrate. The dark circles are due to the formation of lead sulfide on the filter paper. C. Quantification of the image in (B) using the Integrated Density (IntDen) function of the software program ImageJ and subtracting out the background. Notice that this two hour exposure leads to a saturation plateau between the 300 to 400 µg reactions. - After 45 min, take a look at the paper by looking from the underside of the plate. Do not take apart the ‘sandwich’.
- Once strong circles have formed, which usually takes 1 h to 2 h, remove the filter paper and place off to the side. A new filter paper can be placed onto the same plate and the ‘sandwich’ reassembled and placed back into the 37 °C incubator for a second or even third exposure. These secondary exposures will take less time to reach a saturation point, so care must be used to not incubate too long. Approximately 15 min to 30 min is all that is needed to obtain a signal for the secondary/tertiary incubations/exposures when using strong H2S producing tissues like the liver.
- To each finished paper, image with a digital camera or scanner.
Note: Over time, the brown to black circles can become photobleached if exposed to light. Thus, if some samples may have hit a saturation point on the filter paper, the paper can be left out in ambient light for several hours to decrease the intensity of all the bands and bring the circles to a non-saturating point in order to more accurately analyze differences between strong and weak producing samples. However, it is recommended that a new, shorter incubation/exposure be performed (such as the 15-30 min secondary/tertiary exposure) to obtain an image with signals in the linear range. - The image file can then be analyzed by densitometry of each circle via ImageJ using the IntDen (Integrated Density) as the readout. Make sure to also analyze the same size area of a portion of the filter paper where there was no sample to be used for background subtraction.
- The results will give a relative H2S production capacity when compared to a standard, control, or known amount of protein added. It is recommended to run all samples in duplicate or triplicate. Results can be presented as average H2S production capacity relative to a control group or standard, or can be plotted as the raw arbitrary IntDen units given from the ImageJ analysis subtracting the background reading.
- An example of the experimental setup, data output and analysis of the lead acetate/lead sulfide filter paper method and data analysis using a single liver homogenate but with various amounts of protein input (from 0 to 500 µg) is given in Figure 1.
- Prepare lead acetate filter papers
- Agar-embedded lead acetate kinetic assay for H2S production capacity determination from liver extracts and data analysis
This adaptation of the filter-paper embedded lead acetate endpoint assay described above was inspired by the use of a Nafion polymer embedded silver ion microplate for H2S production (Jarosz et al., 2013). This assay offers the potential for kinetic analysis, making it much easier to confirm that the data obtained are in the linear range and below the saturation point. Drawbacks compared to the filter paper method include increased set up time and decreased number of samples that can be run at once. The method is described below:- Prepare the lead acetate agar plate
- Make 50 ml of 1% agarose gel (Agarose HS Molecular Grade from Denville) (see Recipes) in double deionized H2O in a glass Pyrex flask by microwaving until all of the agarose has dissolved.
- Let it cool slightly, but do not let it start solidifying.
- Add lead(II) acetate trihydrate to the agarose to a final concentration of 100 mM. Hand mix the flask until all of the lead(II) acetate trihydrate is dissolved.
Note: Under normal conditions, lead acetate is not volatile under room temperature or in room temperature solutions with water. However, care should be taken, such as wearing a face mask, when putting it into the hot/warm agar. Additionally, proper disposal of lead containing compounds into appropriate chemical waste containers should be completed as per the regulations of each respective research institution. - Pour the 1% agarose/100 mM lead acetate solution onto the top cover of a 96-well culture plate.
- Allow the solution to solidify and cool for 30 min in the dark on a flat surface.
Note: The volume of the agarose/lead acetate solution added to the top cover is approximately 50 ml. However, due to differences in the shapes and styles of 96-well format plates and tops, this volume may need to be adjusted in order to cover the entire surface of the inside of the lid and have enough volume so that the well edges touch the gel in a way that they leave a slight impression into the gel without breaking the gel.
- Make 50 ml of 1% agarose gel (Agarose HS Molecular Grade from Denville) (see Recipes) in double deionized H2O in a glass Pyrex flask by microwaving until all of the agarose has dissolved.
- After the gel has solidified, place it in the dark uncovered while the H2S production capacity reaction solutions and plate are prepared. It should be used immediately and not stored long term in order to prevent drying/cracking of the gel.
- Set up the H2S production assay reactions
- Prepare H2S production capacity reaction solution. This must be freshly done with each assay.
- Calculate the volume needed to run all of the samples. Each sample requires 150 µl of reaction solution.
- In an adequate amount of PBS to cover the needed volume of reaction mixture, add L-cysteine to a final concentration of 10 mM and pyridoxal 5’-phosphate hydrate to a final concentration of 1 mM from stocks as described previously.
- Keep at room temperature and use immediately.
- In each well of a 96-well plate, pipette 150 µl of the reaction mixture.
Note: To prevent cross contamination of signal between wells, it is recommended to skip a space in all directions from each well used. - In each well, pipette 0-300 µg of sample protein.
- Once all samples have been added, ensure there is no residual or spilled liquid on the surface of the 96-well plate by gently wiping with tissue paper.
- Directly place the solidified lead acetate agarose gel containing lid cover over the 96-well plate.
Note: Make sure the lid is firmly in place to avoid cross contamination between wells, but do not press too hard so that the gel is broken. An illustrative description of the assay setup is given in Figure 2A.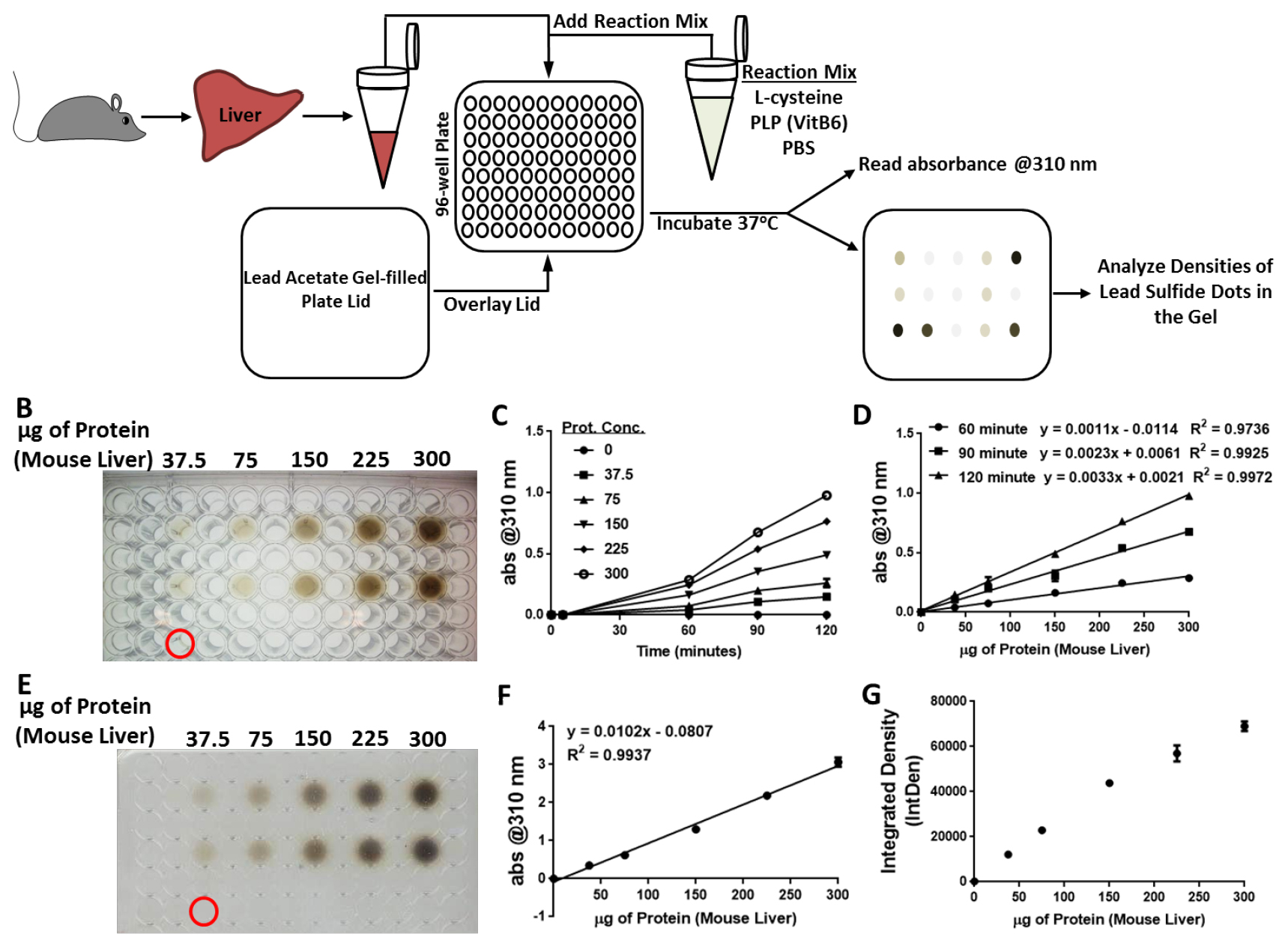
Figure 2. Agar-embedded lead acetate kinetic assay for H2S production capacity determination from liver extracts. A. Schematic overview of the assay. Liver is excised from the mouse, homogenized and lysed in passive lysis buffer followed by protein determination and normalization. In a 96-well plate, the reaction mixture is added first, followed by the liver lysate samples. The lead acetate gel-filled plate lid, which is prepared just prior to setting up the reaction, is placed on top of its compatible 96-well plate. Absorbance at 310 nm is at 0 min to establish a baseline prior to incubating the plate at 37 °C. Throughout the incubation, the absorbance at 310 nm is determined at various time points as lead sulfide gradually forms in the gel. At the completion of the assay, the lead sulfide dots in the gel-filled lid are imaged and analyzed. B. Photographic image of a 96-well plate with a lid cover containing a lead acetate embedded agarose gel after a 2-h incubation at 37 °C. Reaction wells contained various amounts of mouse liver protein (between 0 to 300 µg) as indicated in a 150 µl reaction in PBS containing 10 mM L-cysteine and 1 mM pyridoxal 5’-phosphate hydrate. The dark circles are due to the formation of lead sulfide in the agarose gel. The well highlighted by the red circle contained only the reaction mixture without any protein added and served for background subtraction in the Data analysis. C and D. Plots of the absorbance of light at 310 nm after subtracting background when the entire assembly (plate and lid) was analyzed in the plate reader as a function of time (C) or protein concentration (D) on the x axis. E. Photographic image of the lid cover containing a lead acetate embedded agarose gel after 5 h of incubation at 37 °C. The dark circles are due to the formation of lead sulfide in the agarose gel. The spot highlighted by the red circle corresponds to the well that did not have any protein added and served as background subtraction in the data analysis. F. Plot of the absorbance of light at 310 nm after subtracting background when using just the lid, after a 5 h incubation at 37 °C, was analyzed in the plate reader as a function of protein concentration on the x axis. G. Quantification of the image in (D) using the Integrated Density (IntDen) function of the software program ImageJ and subtracting out the background and plotting the average IntDen for each sample set as a function of protein concentration on the x axis. - Take baseline absorbance at 310 nm reading on the UV-Vis spectrophotometer.
- Incubate the plate at 37 °C in the dark for 0-120 min. At various timepoints between 0-120 min, take the plate out and analyze the absorbance at 310 nm on the plate reader.
- Visual data obtained during the kinetic/timecourse portion of this assay that can be used for endpoint analysis if so desired is done by taking an image of the plate with the lid to visualize the formation of lead sulfide (Figure 2B). Quantitative and numerical data obtained during the kinetic/timecourse portion of this assay is accomplished by subtracting the background absorbance at 310 nm from the 0 µg protein reaction and plotting the absorbance data at 310 nm for the experimental samples. This can be done as a function of time on the x axis (Figure 2C) or as a function of protein concentration on the x axis (Figure 2D).
- Visual data obtained during the endpoint analysis of this assay is done by removing the lid and taking a digital image of the underside of the lid to capture the dark lead sulfide circles formed from H2S exposure to the lead acetate in the gel (Figure 2E). Quantitative and numerical data obtained during the endpoint analysis of this assay is accomplished by placing just the lid upside down into the plate reader and reading the absorbance at 310 nm, subtracting the background, and plotting the values on a graph as a function of protein concentration on the x axis (Figure 2F). Additionally, the digital image of the gel can be analyzed for densitometry of each circle via the software program ImageJ (similar to the procedure described for the lead acetate/lead sulfide filter paper method) using the IntDen (Integrated Density) as the readout. Make sure to also analyze the same size area of a portion of the gel where there was no protein sample added to be used for background subtraction. The values are plotted on a graph as a function of protein concentration on the x axis (Figure 2G).
- Prepare the lead acetate agar plate
Data analysis
- Analysis and interpretation of data obtained in the endpoint assays are as follows:
- Incubate the reaction with the overlaying lead acetate embedded filter paper or agar gel to a point in which detectable but non-saturating lead sulfide circles form.
- Remove the filter paper or agar gel and image with the digital camera or other imaging device, such as a digital image scanner or gel/blot imager.
- The image file can then be analyzed by densitometry of each circle via ImageJ using the IntDen (Integrated Density) as the readout. Make sure to also analyze the same size area of a portion of the filter paper or agar gel where there was no sample to be used for background subtraction.
- The results will give a relative H2S production capacity when compared to a standard, control, or known amount of protein added. It is recommended to run all individual samples in technical duplicates or triplicates and have at least three biological replicates for each condition.
- Results can be presented as average H2S production capacity relative to a control group or standard, or can be plotted as the average raw arbitrary IntDen units given from the ImageJ analysis subtracting the background reading.
- Analysis and interpretation of data obtained in the kinetic assays are as follows:
- Take a baseline absorbance at 310 nm reading on the UV-Vis spectrophotometer.
- Incubate the plate at 37 °C for 0-120 min. At various timepoints between 0-120 min, take the plate out and analyze the absorbance at 310 nm on the plate reader.
- Quantitative and numerical data obtained during the kinetic/timecourse portion of this assay is accomplished by subtracting the background absorbance at 310 nm from the 0 µg protein reaction and plotting the absorbance data at 310 nm for the experimental samples. This can be done as a function of time on the x axis or as a function of protein concentration on the x axis.
- The results will give a relative H2S production capacity when compared to a standard, control, or known amount of protein added at various timepoints. It is recommended to run all individual samples in technical duplicates or triplicates and have at least three biological replicates for each condition.
- Results can be presented as average absorbance at 310 nm for each experimental condition as a function of a specific time or calculated using the area under the curve for all time points.
- With all three of these tests, standard statistical analysis using Student’s t-test can be used when comparing between two groups or an ANOVA with an appropriate multiple comparisons test can be used when comparing values from multiple experimental groups.
Notes
- Although we focus on mammalian liver in this protocol, most of the materials, reagents, equipment and procedures are applicable to other tissue types and organisms. Concentrations of protein, sample volume, L-cysteine concentration, pyridoxal 5’-phosphate hydrate concentration, and incubation times may need to be adjusted dependent on what tissue types and/or organisms are used. We have observed that weaker H2S producing tissues and organisms need longer incubation times (up to 8 h or even overnight), increased protein amounts loaded (up to 1 mg), increased L-cysteine concentrations (up to 100 mM), and/or increased pyridoxal 5’-phosphate hydrate concentrations (up to 10 mM) in order to achieve suitable and quantifiable lead sulfide signals. Additionally, muscle and connective tissue require stronger forms of homogenization than liver, such as the use of bead/bullet homogenization or mechanical knife homogenization. Otherwise, the majority of steps taken in this protocol are the same for both strong and weak H2S producing mammalian tissues. However, tissues/samples from non-mammals, invertebrates, and yeast may need additional and/or alternative steps to obtain optimum results. For example, homogenization of worms (C. elegans) in passive lysis buffer requires more mechanical force via a sonicator or bead/bullet homogenization compared to what is required for liver homogenization and the use of the insoluble/cuticle portion of the worm homogenate can be used in the H2S assay. A recent publication showing a successful C. elegans H2S production capacity assay using the lead acetate to lead sulfide filter paper method can be found here: (Wei and Kenyon, 2016). For fruit flies, whole fly homogenates (including the soluble and insoluble portions) can be used for the H2S assay and normalization is possible by adding the same number of flies (20 to 25 is a suitable number) into the reaction. For yeast, the lead acetate paper can simply be placed on the underside of the plastic/cork/rubber stopper at the top of the culture flask to capture the H2S produced from live yeast cultures.
- Most of the materials, reagents and equipment listed are given with a specific brand and catalog number. This is not meant to be seen as a specific endorsement of that brand and/or product. Many of these can be substituted with other brands and/or catalog numbers that the investigator sees fit to use or with materials that are currently present in the investigator’s lab. Additionally, as time progresses, some of these catalog numbers for certain vendors may change or products may not become available from that vendor, so substitution may be necessary. While we have had success with the proprietary passive lysis buffer (Promega), other non-ionic lysis buffers of known composition could also be used. The lead acetate should be kept away from light and liquid solutions should be made fresh with each use. Gloves and proper personal protective equipment such as safety glasses and lab coat should be worn to protect against contact with the lead and exposure to potential biohazards when dealing with cells and tissues. Proper disposal of lead and cellular and tissue biohazards should be followed as determined by the respective intuition.
- The method of tissue homogenization and lysis was chosen due to its ease of use and ability to maintain proteins in the natural state and preserve enzymatic activity of H2S producing enzymes. However, other methods, such as the use of bead beaters or rotor-stators, could also potentially be used and may be more appropriate for hard-to-homogenize tissues such as muscle and connective tissue.
- To prevent protein degradation and denaturation and to maintain proper enzymatic function, harvest of the liver from mice should be done as soon as possibly post-euthanasia, and all homogenization steps and steps leading up to the enzymatic assay should be done with the protein homogenates/lysates on ice or kept cold (~4 °C to 10 °C).
- Ideal incubation times are always approximate and depend on the amount of protein added as well as the source of the protein, and should stop prior to the saturation point of the lead sulfide spot for the strongest producing sample in that group.
- The amount, in µg, of protein added to each of the reactions is up to the investigator to decide as long as the protein is the limiting factor and not the substrate or co-factor. The more protein added, the quicker the assay will run. However, if too much protein is added, it may cause for the substrate (L-cysteine) or the co-factor (pyridoxal 5’-phosphate hydrate) to become limiting factors, thus compromising the assay and giving false results. In our hands, 100 µg of protein lysate added to the reaction tends to give reliable results.
- L-cysteine is the preferred substrate or ‘fuel’ (Singh et al., 2009) and pyridoxal 5’-phosphate hydrate (PLP) the co-factor (Zhu et al., 2008) for CGL-dependent generation of H2S. L-cysteine working stocks in PBS should be made fresh with each assay. Working stocks of 10 mM PLP in PBS can be prepared ahead of time, aliquoted, and saved at -20 °C for at least several months.
- There are multiple potential substrates for H2S generation by CGL and CBS that could in principle be added used to monitor H2S production capacity. While L-Cys is the preferred substrate for CGL-dependent H2S production, CBS-dependent H2S production condensation of L-Cys together with L-homocysteine is 50x more efficient than L-Cys alone (Chen et al., 2004). Enzyme inhibitors can be further used to distinguish the enzymatic source of H2S. For example, 3 mM propargylglycine, a suicide inhibitor specifically of CGL, can be added prior to the addition of substrate in order to determine the specific contribution of CGL to H2S generation (Kabil et al., 2011).
- Even though the generic cofactor term for what cystathionine beta synthase and cystathionine gamma lyase utilize for their enzymatic activities and production of H2S is called Vitamin B6, it should be noted that for these ex vivo/in vitro H2S assays that only the bioactive form of Vitamin B6 (pyridoxal 5’-phosphate, aka PLP) should be used and not the more common form of Vitamin B6 (pyridoxine).
- A comparison between the two quantification methods (filter paper-based endpoint assay with image-based density quantification vs. agar-based lead acetate method with kinetic absorbance) revealed saturation with high protein concentrations in the endpoint assay but not with the kinetic assay in the given time frame. This could be due to the increased surface for interaction between H2S and lead acetate in the 3-dimensional agar overlay vs. the 2-dimensional filter paper. Additionally, the optical tools for quantification via the plate reader/absorbance method (spectrophotometer and Gen5 analysis software) are more sophisticated than simply analyzing a flat digital image for density. Thus, more information can be extracted from the plate reader/absorbance method than the image/density method at higher protein input levels. However, it seems useful to at least document and analyze the image of the lead sulfide produced on the gel for archival purposes.
- The sensitivity/ limit of detection of the filter paper method (Figure 1) is a function of both protein concentration and incubation/exposure time. Theoretically, if the incubation time is extended (on the order of 4 to 24 h), even a small amount of protein from the liver or other strong H2S producing tissue should give a detectable signal. However, using short incubation/exposure times as described here, we found that even 50 µg of protein gives an adequate signal in the filter paper method. The kinetic agar-based method should theoretically have improved sensitivity over the filter paper method, and here we were able to detect H2S production from 37.5 µg of protein at the 60, 90, and 120 min time points.
Recipes
- 1x passive lysis buffer
8 ml deionized H2O
2 ml 5x passive lysis buffer (Promega) (store at -20 °C)
Make up fresh for each assay and keep on ice during its use
Discard unused 1x passive lysis buffer - 20 mM lead(II) acetate trihydrate
100 ml deionized H2O
759 mg lead(II) acetate trihydrate (Sigma-Aldrich)
Make up fresh at room temperature for each batch of lead acetate filter papers produced - H2S reaction mixture (10 mM L-cysteine, 1 mM pyridoxal 5’-phosphate hydrate, in PBS)
10 ml phosphate buffered saline (aka PBS), without calcium and without magnesium, pH 7.4 (Fisher Scientific)
11.9 mM phosphates
137 mM sodium chloride
2.7 mM potassium chloride
12.1 mg L-cysteine (Sigma-Aldrich)
2.5 mg pyridoxal 5’-phosphate hydrate (Sigma-Aldrich)
Mix all reagents in a 15 ml centrifuge tube using a vortex mixer
Notes: - Not all of the pyridoxal 5’-phosphate hydrate (PLP) will go into solution. After you have mixed for 1-2 min, briefly spin down in a centrifuge to pellet the undissolved PLP and save the supernatant for use.
- This must be made fresh for each assay. Once made, use immediately and do not put on ice
- 1% agarose gel with 100 mM lead(II) acetate trihydrate
- 50 ml double deionized H2O
500 mg HS molecular biology grade agarose (Denville)
Mix in a large flask and microwave until all of the agar is dissolved
Let it cool slightly, but do not let it start solidifying - Add 1.9 g lead(II) acetate trihydrate (Sigma-Aldrich) to the agarose
- Hand mix the flask until all of the lead(II) acetate trihydrate is dissolved. Make sure to do this quickly before the agarose solidifies
- Pour the 1% agarose/100 mM lead acetate solution onto the top cover of a 96-well culture plate and let it cool and solidify in the dark on a flat surface
Note: This must be done fresh just prior to running the assay.
Acknowledgments
The authors thank Marissa Granitto for testing of the protocols and providing critical feedback on the manuscript. This work was supported by grants and funds provided by DK090629 and AG036712 (JRM), and AG050777 (CH). The authors declare that there are no conflicts of interests or competing interests related to the design and implementation of this protocol.
References
- Bhatia, M., Wong, F. L., Fu, D., Lau, H. Y., Moochhala, S. M. and Moore, P. K. (2005). Role of hydrogen sulfide in acute pancreatitis and associated lung injury. FASEB J 19(6): 623-625.
- Chen, X., Jhee, K. H., and Kruger, W. D. (2004). Production of the neuromodulator H2S by cystathionine β-synthase via the condensation of cysteine and homocysteine. J Biol Chem 279: 52082-52086.
- Dickhout, J. G., Carlisle, R. E., Jerome, D. E., Mohammed-Ali, Z., Jiang, H., Yang, G., Mani, S., Garg, S. K., Banerjee, R., Kaufman, R. J., Maclean, K. N., Wang, R. and Austin, R. C. (2012). Integrated stress response modulates cellular redox state via induction of cystathionine γ-lyase: cross-talk between integrated stress response and thiol metabolism. J Biol Chem 287(10): 7603-7614.
- Hartle, M. D., and Pluth, M. D. (2016). A practical guide to working with H2S at the interface of chemistry and biology. Chem Soc Rev 45: 6108-6117.
- Hine, C., Harputlugil, E., Zhang, Y., Ruckenstuhl, C., Lee, B. C., Brace, L., Longchamp, A., Trevino-Villarreal, J. H., Mejia, P., Ozaki, C. K., Wang, R., Gladyshev, V. N., Madeo, F., Mair, W. B. and Mitchell, J. R. (2015). Endogenous hydrogen sulfide production is essential for dietary restriction benefits. Cell 160(1-2): 132-144.
- Ikeda, M., Ishima, Y., Shibata, A., Chuang, V. T. G., Sawa, T., Ihara, H., Watanabe, H., Xian, M., Ouchi, Y., Shimizu, T., Ando, H., Ukawa, M., Ishida, T., Akaike, T., Otagiri, M. and Maruyama, T. (2017). Quantitative determination of polysulfide in albumins, plasma proteins and biological fluid samples using a novel combined assays approach. Anal Chim Acta 969: 18-25.
- Jarosz, A. P., Yep, T. and Mutus, B. (2013). Microplate-based colorimetric detection of free hydrogen sulfide. Anal Chem 85(7): 3638-3643.
- Kabil, O., Vitvitsky, V., Xie, P. and Banerjee, R. (2011). The quantitative significance of the transsulfuration enzymes for H2S production in murine tissues. Antioxid Redox Signal 15(2): 363-372.
- Kuester, E. and Williams, S. T. (1964). Production of hydrogen sulfide by Streptomycetes and methods for its detection. Appl Microbiol 12, 46-52.
- McBride, R. S. and Edwards, J. D. (1914). Lead acetate test for hydrogen sulphide in gas. United States Department of Commerce 41: 4-46.
- Mitchell, S. J., Madrigal-Matute, J., Scheibye-Knudsen, M., Fang, E., Aon, M., Gonzalez-Reyes, J. A., Cortassa, S., Kaushik, S., Gonzalez-Freire, M., Patel, B., Wahl, D., Ali, A., Calvo-Rubio, M., Buron, M. I., Guiterrez, V., Ward, T. M., Palacios, H. H., Cai, H., Frederick, D. W., Hine, C., Broeskamp, F., Habering, L., Dawson, J., Beasley, T. M., Wan, J., Ikeno, Y., Hubbard, G., Becker, K. G., Zhang, Y., Bohr, V. A., Longo, D. L., Navas, P., Ferrucci, L., Sinclair, D. A., Cohen, P., Egan, J. M., Mitchell, J. R., Baur, J. A., Allison, D. B., Anson, R. M., Villalba, J. M., Madeo, F., Cuervo, A. M., Pearson, K. J., Ingram, D. K., Bernier, M. and de Cabo, R. (2016). Effects of sex, strain, and energy intake on hallmarks of aging in mice. Cell Metab 23(6): 1093-1112.
- Módis, K., Coletta, C., Erdelyi, K., Papapetropoulos, A. and Szabo, C. (2013). Intramitochondrial hydrogen sulfide production by 3-mercaptopyruvate sulfurtransferase maintains mitochondrial electron flow and supports cellular bioenergetics. FASEB J 27(2): 601-611.
- Nikonorova, I. A., Al-Baghdadi, R. J., Mirek, E. T., Wang, Y., Goudie, M. P., Wetstein, B. B., Dixon, J. L., Hine, C., Mitchell, J. R., Adams, C. M., Wek, R. C. and Anthony, T. G. (2017). Obesity challenges the hepatoprotective function of the integrated stress response to asparaginase exposure in mice. J Biol Chem.
- Olson, K. R. (2009). Is hydrogen sulfide a circulating "gasotransmitter" in vertebrate blood? Biochim Biophys Acta 1787, 856-863.
- Olson, K. R. (2012). A practical look at the chemistry and biology of hydrogen sulfide. Antioxid Redox Signal 17(1): 32-44.
- Polhemus, D. J., Calvert, J. W., Butler, J. and Lefer, D. J. (2014). The cardioprotective actions of hydrogen sulfide in acute myocardial infarction and heart failure. Scientifica (Cairo) 2014: 768607.
- Singh, S., Padovani, D., Leslie, R. A., Chiku, T. and Banerjee, R. (2009). Relative contributions of cystathionine β-synthase and γ-cystathionase to H2S biogenesis via alternative trans-sulfuration reactions. J Biol Chem 284(33): 22457-22466.
- Stipanuk, M. H. and Beck, P. W. (1982). Characterization of the enzymic capacity for cysteine desulphhydration in liver and kidney of the rat. Biochem J 206(2): 267-277.
- Takano, Y., Shimamoto, K. and Hanaoka, K. (2016). Chemical tools for the study of hydrogen sulfide (H2S) and sulfane sulfur and their applications to biological studies. J Clin Biochem Nutr 58(1): 7-15.
- Wang, R. (2012). Physiological implications of hydrogen sulfide: a whiff exploration that blossomed. Physiol Rev 92(2): 791-896.
- Wei, Y. and Kenyon, C. (2016). Roles for ROS and hydrogen sulfide in the longevity response to germline loss in Caenorhabditis elegans. Proc Natl Acad Sci U S A 113(20): E2832-2841.
- Yang, G., Wu, L., Jiang, B., Yang, W., Qi, J., Cao, K., Meng, Q., Mustafa, A.K., Mu, W., Zhang, S., Snyder, S. H., Wang, R. (2008). H2S as a physiologic vasorelaxant: hypertension in mice with deletion of cystathionine γ-lyase. Science 322, 587-590.
- Zhang, Y. and Weiner, J. H. (2014). A simple semi-quantitative in vivo method using H2S detection to monitor sulfide metabolizing enzymes. Biotechniques 57(4): 208-210.
- Zhu, W., Lin, A. and Banerjee, R. (2008). Kinetic properties of polymorphic variants and pathogenic mutants in human cystathionine γ-lyase. Biochemistry 47(23): 6226-6232.
Article Information
Copyright
© 2017 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Hine, C. and Mitchell, J. R. (2017). Endpoint or Kinetic Measurement of Hydrogen Sulfide Production Capacity in Tissue Extracts. Bio-protocol 7(13): e2382. DOI: 10.21769/BioProtoc.2382.
Category
Cell Biology > Tissue analysis > Tissue isolation
Cell Biology > Cell signaling > Second messenger
Biochemistry > Other compound > Hydrogen sulfide
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.