- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Affinity Purification of the RNA Degradation Complex, the Exosome, from HEK-293 Cells

Published: Vol 7, Iss 8, Apr 20, 2017 DOI: 10.21769/BioProtoc.2238 Views: 10036
Reviewed by: Gal HaimovichAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Jul 2016
Advertisement




Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols


Abstract
The RNA exosome complex plays a central role in RNA processing and regulated turnover. Present both in cytoplasm and nucleus, the exosome functions through associations with ribonucleases and various adapter proteins (reviewed in [Kilchert et al., 2016]). The following protocol describes an approach to purify RNA exosome complexes from HEK-293 cells, making use of inducible ectopic expression, affinity capture, and rate-zonal centrifugation. The obtained RNA exosomes have been used successfully for proteomic, structural, and enzymatic studies (Domanski et al., 2016).
Keywords: RNA exosomeBackground
In our previous work, we established an isogenic HEK-293 cell line expressing C-terminally 3xFLAG-tagged exosome component EXOSC10 (RRP6) under the control of a tetracycline-inducible CMV promotor (HEK-293 Flp-In T-REx – Thermo Fisher Scientific). This system permitted us to express the tagged EXOSC10 protein at a level comparable to the endogenous WT protein, and to explore exosome purification protocols using a magnetic anti-FLAG affinity medium and protein extracts derived from cryomilled cell powder (Domanski et al., 2012). Further exploring the protein extraction conditions used, we developed a protocol permitting the retention of DIS3 (RRP44) within affinity captured exosomes, which has otherwise proven difficult (Hakhverdyan et al., 2015). Building on these studies, we further purified RNA exosomes, +/- DIS3, by rate-zonal centrifugation using glycerol density gradients (Domanski et al., 2016). Although the presence of the detergent CHAPS (3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate) in the protein extract enhanced the yield of DIS3 co-purifying with affinity captured exosomes, the interaction was subsequently lost during sedimentation in a glycerol density gradient. To counteract this, the crosslinker DTSSP [3,3’-dithiobis(sulfosuccinimidyl propionate)] was employed. The treatment enabled the retention of DIS3 within exosomes during sedimentation, but negatively affected DIS3 enzymatic functions. The peak fractions from DIS3 +/- exosome fractions both contained apparent exoribonucleolytic activities consistent with EXOSC10-derived distributive 3’-5’ hydrolysis. Apparent structural differences between samples that retained DIS3 (DTSSP-treated) and those that did not could be observed by negative stain electron microscopy. The protocol presented here will enable users to obtain endogenously assembled RNA exosome fractions suitable for additional analytical methods including in vitro biochemistry, enzymology, and electron microscopy. Note that many aspects of this protocol can be easily adapted, e.g., to use (1) different affinity tags and expression contexts, or (2) antibodies against the endogenous protein (LaCava et al., 2015).
Materials and Reagents
Note: Catalog numbers are given for most of the reagents listed below; an equivalent quality reagent from an alternative supplier can typically be substituted with comparable results. Standard materials and reagents for mammalian cell culture are required and are not all explicitly listed below.
- Pipette tips
- Nunclon cell culture 245 mm (500 cm2) square dish (Sigma-Aldrich, catalog number: D8679 )
- Nunclon 175 cm2 cell culture flask (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 156502 )
- 15” cell scraper (Fisher Scientific, catalog number: 08-100-242 )
- 50 ml polypropylene conical tubes (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 339652 )
- 20 ml Luer-lock syringe (BD, catalog number: 302830 )
- Parafilm (Sigma-Aldrich, catalog number: P7793 )
- Syringe end caps (Bio-Rad Laboratories, catalog number: 7311660EDU )
- 2 ml microcentrifuge tubes (e.g., Eppendorf, catalog number: 022363344 )
- 0.45 μm polyethersulfone (PES) sterile syringe filters (VWR, catalog number: 28145-505 )
- 5 ml Ultracentrifuge tubes (Seton Scientific, catalog number: 7022 )
Note: If you will use the BioComp Instruments Piston Gradient Fractionator to recover fractions of purified exosomes (see Equipment), we recommend you obtain these tubes from BioComp because they are tolerance tested for compatibility. - HEK-293 Flp-In T-REx EXOSC10-3xFLAG cells ([Domanski et al., 2016]; available upon request)
- DMEM, high glucose, GlutaMAX (Thermo Fisher Scientific, catalog number: 31966047 )
- Fetal bovine serum (FBS), tetracycline-free
Note: Numerous suppliers can provide this. Many suppliers carry FBS products not labelled as tetracycline-free, but consulting the product specification sheet for a given lot may reveal that tetracycline has been tested for and found to be absent. In our hands, the performance of such lots has been identical to ‘certified’ tetracycline-free FBS. - 100x penicillin-streptomycin (P/S) (Thermo Fisher Scientific, GibcoTM, catalog number: 15140122 )
- Trypsin-EDTA, 0.05% (Thermo Fisher Scientific, GibcoTM, catalog number: 25300054 )
- Tetracycline (Sigma-Aldrich, catalog number: 87128 )
Note: Prepare a stock solution of 10 mg/ml in ethanol and store at -20 °C. The working solution is 5 μg/ml–for the induction add 1 μl per 1 ml cell culture medium. Doxycycline can also be used. - Phosphate-buffered saline (PBS), pH 7.4 (Thermo Fisher Scientific, catalog number: 10010023 )
- 100x L-glutamine (200 mM) (Thermo Fisher Scientific, GibcoTM, catalog number: 25030081 )
- TrypLE dissociation reagent (Thermo Fisher Scientific, GibcoTM, catalog number: 12605010 )
- Phenol red solution, 0.5% (Sigma-Aldrich, catalog number: P0290 )
- Liquid nitrogen (LN2)
- Freestyle 293 medium (Thermo Fisher Scientific, GibcoTM, catalog number: 12338026 )
- 3 M ammonium sulfate in 0.1 M sodium phosphate buffer pH 7.4
- Bovine serum albumin (BSA) (New England Biolabs, catalog number: B9000S )
- Glycerol (Sigma-Aldrich, catalog number: G5516 )
- 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid (HEPES) (Sigma-Aldrich, catalog number: 54457 )
- Sodium chloride (NaCl) (Sigma-Aldrich, catalog number: S3014 )
- Sodium hydroxide (NaOH) (Sigma-Aldrich, catalog number: 71687 )
- Hydrochloric acid (HCl) (Sigma-Aldrich, catalog number: 320331 )
- Triton X-100 (Sigma-Aldrich, catalog number: T8787 )
- Protease inhibitor cocktail, EDTA-free (Roche Diagnostics, catalog number: 11873580001 )
- CHAPS (Sigma-Aldrich, catalog number: C5070 )
- Anti-FLAG M2 antibody (Sigma-Aldrich, catalog number: F3165 or F1804 )
- Dynabeads M-270 epoxy (Thermo Fisher Scientific, InvitrogenTM, catalog number: 14302D )
- 0.1 M sodium phosphate buffer pH 7.4
- Tris base (Sigma-Aldrich, catalog number: 93362 )
- 3xFLAG peptide (Sigma-Aldrich, catalog number: F4799 ) reconstituted at 5 mg/ml in TBS (50 mM Tris-HCl pH 7.4, 150 mM NaCl). Aliquots should be stored at -20 °C
- DTSSP (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 21578 )
- 4x LDS (lithium dodecyl sulfate) sample loading buffer (Thermo Fisher Scientific, NovexTM, catalog number: NP0007 )
- 10x sample reducing agent (Thermo Fisher Scientific, NovexTM, catalog number: NP0004 ); or 500 mM dithiothreitol (DTT)
- 4-12% Bis-Tris 26-well midi gels (Thermo Fisher Scientific, InvitrogenTM, catalog number: WG1403BOX )
- 20x MOPS (3-morpholinopropane-1-sulfonic acid) gel running buffer (Thermo Fisher Scientific, NovexTM, catalog number: NP0001 )
- SilverQuest Silver Staining Kit (Thermo Fisher Scientific, NovexTM, catalog number: LC6070 )
- Sypro ruby protein gel stain (Sigma-Aldrich, catalog number: S4942 )
- Anti-EXOSC10 antibody (Abcam, catalog number: ab95028 )
- Anti-SKIV2L2 antibody (Abcam, catalog number: ab70552 )
- Anti-EXOSC3 antibody (Proteintech, catalog number: 15062-1-AP )
- ExoI extraction solution (see Recipes)
- ExoII extraction solution (see Recipes)
- Gradient solution – ‘light’ (see Recipes)
- Gradient solution – ‘heavy’ (see Recipes)
Equipment
Note: Catalog numbers are given for most of the equipment listed below; instruments from alternative manufacturers may be substituted provided equivalent functionality.
- Pipettes
- Balance
- CO2 incubator for mammalian cell culture
- Refrigerated microcentrifuge (capable of reaching 20,000 x g)
- New Brunswick Innova 2000 platform shaker (Eppendorf, New BrunswickTM, model: Innova® 2000 , catalog number: M1190-0002)
Note: Any shaker installed in a mammalian cell culture incubator must be able to tolerate continuous high humidity (~90% relative humidity). - 250 ml plastic beaker
- Hemocytometer
- 1 L square PYREX bottles (Corning, PYREX®, catalog number: 1396-1L )
- Milling balls, stainless steel, 20 mm (Retsch, catalog number: 05.368.0062 )
- 50 ml stainless steel milling jar (Retsch, catalog number: 01.462.0149 )
- Metal spatula
- Planetary Ball Mill PM 100 (Retsch, model: PM 100 , catalog number: 20.540.0001)
- Thermomixer (Eppendorf, model: Thermomixer® R , catalog number: 5355000.011; or equivalent)
- Vortex mixer equipped with head for multiple 1.5/2.0 ml tubes (Thermo Fisher Scientific, Fisher Scientific, model: Fisher ScientificTM Vortex Mixer , catalog number: 02-215-386)
- Neodymium magnet microfuge tube rack (Thermo Fisher Scientific, catalog number: 12321D )
- Ultracentrifuge compatible with either of the rotors listed below (Beckman Coulter), e.g., Optima L or Optima MAX series (Beckman Coulter, model: Optima L or Optima MAX series )
- SW 55 Ti or MLS-50 rotor (Beckman Coulter, model: SW 55 Ti or MLS-50 )
- Microtip sonicator (e.g., Qsonica, model: Q700 ) equipped with a low intensity 1/16” microtip probe (Qsonica, catalog number: 4717)
- Gradient fractionator and accessories for SW 55 Ti (BioComp Instruments, catalog number: 152-001 )
- Gradient master and accessories for SW 55 Ti (BioComp Instruments, catalog number: 107-201M )
Procedure
Note: Many of the steps described below for harvesting cells, milling them to powder, and carrying out an affinity capture can be viewed in our online video protocol (LaCava et al., 2016).
- Cell culturing and harvesting
The tetracycline inducible EXOSC10-3xFLAG HEK-293 cell line can be obtained by contacting the authors or by following the manufacturer’s instructions. Except where noted, standard mammalian cell culturing procedures apply (Freshney 2011; Uphoff and Drexler, 2013).- Adherent cell growth
- Seed ~107 cells on each of 16 x 500 cm2 square dishes in 90 ml DMEM supplemented with 10% v/v FBS and 1x P/S (DMEM-FBS-P/S).
Note: 16 x 500 cm2 square dishes should yield at least 10 g of cell pellet, wet cell weight (WCW). - When the cells reach ~90% confluency, remove the medium and induce the expression of EXOSC10-3xFLAG by adding fresh DMEM-FBS-P/S supplemented with 5 ng/ml tetracycline (Tet).
- Harvest the cells at 24 h post induction by scraping in ice-cold PBS–we use 30 ml/plate.
Note: Alternatively, the cells can be harvested by trypsinization. For each plate, wash the cells with 20 ml PBS, add 10 ml of 0.05% trypsin-EDTA, tilt the dish to expose the cell monolayer, remove the excess liquid, and place them back into the CO2 incubator for ~5 min. Resuspend the cells in 10 ml DMEM-FBS-P/S and transfer to a 50 ml tube. - Centrifuge the harvested cells at 1,000 x g for 5 min (4 °C).
- Discard the supernatant, resuspend, and combine the pellets in ice-cold PBS (~100 ml total volume), within two 50 ml tubes.
- Centrifuge at 1,000 x g for 5 min (4 °C).
- Discard the supernatant, resuspend the pellets in ice-cold PBS (~1 volume of PBS per volume of the pellet), and transfer the solution to a 20 ml capped syringe. Seal upper opening of the syringe with Parafilm (to avoid any accidental spillage) and place it inside a 50 ml tube.
- Centrifuge at 1,000 x g for 5 min (4 °C).
- Remove any remaining liquid, replace the plunger, and carefully inject the content into LN2.
Note: First, fill 250 ml plastic beaker (placed inside a Styrofoam box) with LN2, then inject the cells directly into LN2. - Store at -80 °C prior to cryomilling.
- Seed ~107 cells on each of 16 x 500 cm2 square dishes in 90 ml DMEM supplemented with 10% v/v FBS and 1x P/S (DMEM-FBS-P/S).
- Suspension cell growth
For this protocol you will need a platform shaker with appropriate tolerances, installed inside of a 37 °C, humidified, 8% CO2 incubator. Suspension cell growth offers a convenient and cost effective way to generate large quantities of cells and maintain continuous cultures. The protocol presented here is adapted from a method we previously adapted (Muller et al., 2005) and applied to HEK-293TLD cells (Dai et al., 2012; Taylor et al., 2013; Taylor et al., 2016). Using HEK-293 Flp-In T-REx cells, suspension growth may yield ~3-5 million cells per ml, with growth slowing above ~3 million/ml. Obtaining accurate cell counts can be challenging because these suspensions tend to grow in clumps and require trituration by pipetting to be counted in a hemocytometer. Yields in WCW should be ≥ 4 g per 400 ml culture of cells expressing 3xFLAG-tagged EXOSC10 at near the endogenous level. - Seed cells from stocks in the standard way and obtain 2 x 175 cm2 flasks of nearly confluent (90%+) cells. We use 25 ml of medium per 175 cm2 flask.
- Split the cells into 4 x 175 cm2 flasks in a 1:1 mixture of DMEM-FBS-P/S and Freestyle medium supplemented with 2% v/v FBS, 2 mM L-glutamine, and 0.5x P/S (the latter, referred to as conditioning medium). This initiates conditioning of adherent cells for suspension growth.
Note: We have been unable to inoculate new suspension cultures directly from frozen cell stocks of suspension conditioned HEK-293 Flp-In T-REx cells. Therefore, we freshly condition cells for each new suspension growth, and then maintain cells in suspension for the needed duration. The difficulty in inoculating directly to suspension and the lower maximal cell density observed for HEK-293 Flp-In T-REx cells compared to HEK-293TLD cells may be due to the absence of the SV40 large T antigen in the former (Ahuja et al., 2005; Taylor et al., 2016). - At the next cell splitting use a 1:3 mixture of DMEM-FBS-P/S and conditioning medium, transfer the cells into 8 x 175 cm2 flasks and grow to near confluence.
- Wash the cells with 10 ml PBS and discard the wash, then release the cells from the flasks using 0.1x TrypLE (1:10 dilution in PBS). Add 1.5 ml of 0.1x TrypLE solution, tilt each flask to expose the cell monolayer, and then remove the excess liquid. Incubate the flasks at RT for 10 min.
- Resuspend the released cells from each flask in 10 ml of conditioning medium. Combine 40 ml of the cell suspension and 60 ml of conditioning medium supplemented with 200 μl of 0.5% w/v phenol red (final 0.001% w/v) in a 1 L square glass bottle. This typically results in two 100 ml suspension cultures at a cell density of around a couple million per ml. Suspension cultures should be inoculated at between ~0.5-3 million cells/ml. Leave the bottle caps one-full-turn open (from just-closed) and set the cultures shaking at 130 RPM within the incubator.
Note: The phenol red (PR) functions as a pH indicator and an indirect method of monitoring cell density in culture, complementing cell counting. The solution should have a light red appearance similar to commercially available DMEM, which also frequently contains this additive. The working volume for 1 L glass bottles in this protocol is between 100-400 ml. Two 400 ml cultures should ultimately yield at least 8 g WCW. - Monitor the culture by cell counting and medium color. When the cell density is very near or exceeds 3 million cells/ml and begins to lighten in color towards orange, dilute the culture to 1-2 million cells/ml with 100 ml of Freestyle medium supplemented with 2 mM L-glutamine, 0.5x P/S, and 0.001% PR (no FBS)–resulting in a 1% final FBS concentration while other reagent concentrations remain the same (referred to as suspension medium). All future dilutions can be made with suspension medium.
Note: Cells are typically round, single, and relatively easy to count for a few days after the initial inoculation, and then will become increasingly clumpy, even adhering to some extent to the side of the flask where the liquid line reaches. - Continue to monitor the culture by cell counting and medium color. When the cell density is very near or exceeds 3 million cells/ml and begins to lighten in color towards orange, dilute the cultures to ≤ 3 million cells/ml with 200 ml suspension medium.
- Once this 400 ml of medium is very near or exceeds 3 million cells/ml, remove 50 ml of the culture from each and use this to seed two or more new 100 ml cultures at ~0.5-2 million cells/ml (if desired). To the remaining 350 ml of culture, add 50 ml of suspension medium supplemented with 8 ng/ml Tet (1 ng/ml final), and incubate overnight (~16-24 h).
Note: We observed that 1 ng/ml of tetracycline in suspension culture was sufficient to give expression comparable to 5 ng/ml in adherent growth. However, we encourage researchers to test expression in their hands by carrying out an anti-RRP6 Western blot after induction (Domanski et al., 2012). This protocol can be miniaturized using smaller culture vessels for pre-tests of that nature. - Harvest the cells by centrifugation, wash and freeze in LN2 as described in steps d-j in section A1–Adherent cell growth–above.
- Adherent cell growth
- Cryomilling procedure
The cryogenic disruption of the mammalian cell pellets has been previously described (Domanski et al., 2012; Taylor et al., 2016; LaCava et al., 2016). Briefly, pre-cool a milling jar, 2 x 20 mm grinding balls, a metal spatula, and a 50 ml tube by immersing in LN2. Remove any LN2 from the jar chamber and place the cell pellets inside. Set the counter balance and clamp the jar in the Retsch PM 100. Run the machine for 3 milling cycles of 3 min each (reverse rotation, 1 min interval, no break time) at 400 RPM. Cool down the milling jar with LN2 between cycles. Recover the resulting cell powder using a spatula and transfer to 50 ml tube. Store at -80 °C. - Coupling of the anti-FLAG M2 antibodies to Dynabeads M-270 epoxy
The production of anti-FLAG magnetic medium has been previously described in detail (Cristea and Chait, 2011; Domanski et al., 2012; Taylor et al., 2016) and can also be carried out according to the manufacturer’s instructions (Thermo Fisher Scientific) with comparable results. Briefly, prepare 20 μl of 0.5 μg/μl antibody, in a sodium phosphate (pH 7.4) buffered 1 M ammonium sulfate solution, per mg of magnetic beads to be antibody-conjugated (i.e., 10 μg of antibody, in 20 μl solution, will be used per mg of beads). Combine the antibody solution and the magnetic beads and incubate overnight (16-24 h) at 30-37 °C with rotation. The mixing applied should be sufficient to ensure the beads remain in suspension for the duration of the incubation. For small batches, conjugation can be performed at 37 °C in a thermomixer (≥ 1,200 RPM). After coupling, remove the excess antibody solution, wash the beads to remove residual uncoupled antibodies, and finally resuspend the antibody-conjugated magnetic medium in 1x PBS [final] containing 0.5 mg/ml BSA and 50% v/v glycerol (storage solution): add 6.7 μl of storage solution per 1 mg of medium (resulting in a slurry of ~15% w/v) and store at -20 °C. The slurry can be stored in this way for at least 1 year without any noticeable loss of performance. - Affinity purification
A general procedure describing and demonstrating best practices for affinity purification using mammalian cell powder and magnetic media has been previously described (LaCava et al., 2016). The following implementation has been optimized for obtaining human RNA exosomes. Here, DIS3- exosomes are referred to as ExoI and the DIS3+ exosomes are ExoII.- Pre-cool a metal spatula and 4 x 2 ml safe-lock tubes in LN2.
- Weight out 4 x 250 mg of HEK-293 EXOSC10-3xFLAG cell powder.
Note: It is important to keep the cell powder frozen. Leave the tubes in LN2 until the next step is initiated. - Place the samples in the rack, open the caps, and let them stand at room temperature for 1 min.
- Add 1,250 μl ExoI or ExoII solution to each (see Recipes).
Note: ExoI solution yields DIS3- exosomes, whereas ExoII yields DIS3+ exosomes. - Vortex at maximum speed to fully resuspend the cell powder (should not require more than 30 sec), and immediately place the samples on ice.
Note: Once the cell powder is resuspended, the sample should be held on ice between all manipulations throughout the procedure unless otherwise stated. - Briefly sonicate to completely disperse and homogenize the cell powder. Use 4 pulses, 2 sec each (~30 J total per 250 mg sample) (LaCava et al., 2016).
Note: If clumps are visible, the sonication step should be repeated until no clumps are obvious by visual inspection. - Clarify the extract by centrifugation at 20,000 x g for 10 min (4 °C).
- While the samples are in the centrifuge, distribute 4x 25 μl anti-FLAG beads slurry into 2 ml microcentrifuge tubes and wash twice with 1 ml of the extraction solution of choice to thoroughly remove the storage solution.
Note: We use 10 μl of the beads slurry per 100 mg of the cell powder. To wash the beads, combine 1 ml of extraction solution with 25 μl anti-FLAG magnetic medium (slurry) in a 2 ml microcentrifuge tube and vortex briefly to fully resuspend the beads. Pulse-spin the tube briefly in a mini-centrifuge to collect all the solution at the bottom and then place the tube in a magnetic tube rack until the beads are collected at the side of the tube. Remove the supernatant using a pipet or an aspirator, and repeat the washing step one more time. Perform both washing steps at room temperature. After washing, the beads may be held on ice and are ready for use. Alternatively, one can also wash a total of 100 μl anti-FLAG medium and distribute across 4 tubes after washing. - Combine the clarified extracts with the pre-washed beads. Incubate for 1 h with rotation at 4 °C (cold room).
- Collect the beads on a magnet, remove the supernatant, and wash with 1 ml of ice cold extraction solution, in the manner noted above.
- Wash with extraction solution two more times. Move each sample to a fresh tube during the second wash.
Note: Moving each sample (beads resuspended in extraction solution) to a fresh tube minimizes contamination of the eluate. Cell extract proteins non-specifically adsorbed to the internal surfaces of the tube used for affinity capture may be released into the sample during subsequent manipulations. - After the 3rd wash, spin the samples briefly in a mini-centrifuge.
- Place again on a magnet and carefully remove any remaining liquid.
- Add 30 μl of elution solution (1 mg/ml 3xFLAG peptide diluted from the stock in extraction solution) to each tube.
- Incubate the samples at room temperature for 15 min with gentle agitation. ~60% power on a vortex equipped with a head for multiple 1.5/2.0 ml tubes.
Note: Make sure the beads are not being mixed too vigorously, nor settling at the bottom of the tube. - Place on a magnet and collect the supernatants.
- Add 30 μl of the extraction solution to wash the beads. Collect as above and pool the fractions together.
- Keep the samples on ice prior to loading on a gradient.
- Pre-cool a metal spatula and 4 x 2 ml safe-lock tubes in LN2.
- DTSSP crosslinking to obtain ExoII
DIS3 can be co-purified with the exosome in the ExoII extraction solution. However, it dissociates during sedimentation in a glycerol gradient. To preserve this interaction, we used chemical crosslinking. DTSSP will enhance the preservation of the DIS3/exosome interaction, but DIS3 enzymatic activity will be compromised by this treatment.- Add 2 μl of freshly prepared 1.2 mM DTSSP per 10 μl of ExoII prepared by elution with 3xFLAG as described above.
- Carry out the crosslinking reaction for 50 min at RT.
- Quench by adding 2 μl 1 M Tris-HCl pH 8.
- Keep on ice prior to loading on a gradient.
- Add 2 μl of freshly prepared 1.2 mM DTSSP per 10 μl of ExoII prepared by elution with 3xFLAG as described above.
- Rate zonal centrifugation
The following settings and parameters are optimized for an SW 55 Ti rotor in a Beckman Optima L series ultracentrifuge. For MLS-50 details, refer to (Domanski et al., 2016).- Prepare a 10-40% glycerol gradient using the BioComp Gradient Master instrument (short cap, 10-40% glycerol v/v) or other appropriate method. Always prepare an extra gradient to use as a balance.
- Pre-cool formed gradient to 4 °C (typically ≥ 1 h in a cold room).
- Load the sample(s) by gently pipetting onto the top of the gradient.
Note: To avoid disrupting the top layer of the gradient, rest the pipette tip against the wall of the tube, just above the meniscus of the glycerol solution, and slowly eject the sample. - Run the gradient using the following settings: minimum acceleration; no brake; 4 °C; 50k RPM; 6 h 36 min.
- After the run is completed, collect the fractions using a BioComp Piston Gradient Fractionator (consult the manufacturer’s instructions), or other appropriate method. We used the following settings with manual fraction collection: distance–2 mm, speed–0.3 mm/sec. This collection regime will produce a meniscus fraction, ~20 fractions of ~225 μl, and a bottom fraction. Some variations will be observed depending on the specific set-up implemented, however, using the described procedure and setup we retrieved the peak of sedimented ExoI in fraction 11 and ExoII in fractions 11 and 12.
- Prepare a 10-40% glycerol gradient using the BioComp Gradient Master instrument (short cap, 10-40% glycerol v/v) or other appropriate method. Always prepare an extra gradient to use as a balance.
- SDS-PAGE analysis of gradient fractions
- Mix 14 μl of the gradient fraction with 5 μl of 4x LDS and 1 μl of 10x reducing agent (or 500 mM DTT); heat at 70 °C for 10 min for ExoI (DIS3-) fractions or to 75 °C for 20 min for DTSSP-treated ExoII (DIS3+) fractions.
- Load the samples on a 26-well, 4-12% NuPAGE Bis-Tris gel, following the manufacturer’s instructions.
- Run at 200 V until the tracking dye reaches the bottom of the gel cassette.
- Remove the gel from the plastic cassette and place in a clean container.
- Stain with the silver or Sypro Ruby following manufacturers’ instructions.
- Mix 14 μl of the gradient fraction with 5 μl of 4x LDS and 1 μl of 10x reducing agent (or 500 mM DTT); heat at 70 °C for 10 min for ExoI (DIS3-) fractions or to 75 °C for 20 min for DTSSP-treated ExoII (DIS3+) fractions.
Data analysis
During the sedimentation, free proteins (or partially assembled complexes) can be found close to the top of the gradient, whereas intact RNA exosomes migrate further and can be found around the middle of a gradient. Fractions containing intact exosomes exhibit signature staining intensities for (from the top of the gel) SKIV2L2, EXOSC10-3xFLAG, EXOSC9 and the core/low mass proteins, approximately distributed at the expected molecular masses (EXOSC9 runs closer to 60 kDa than its predicted mass of ~49 kDa). Figure 1A depicts a stained SDS-polyacrylamide gel loaded with gradient fractions obtained after ultracentrifugation (real data shown in Figure 1B). In this example, fraction number eleven (11) demonstrates the composition and staining intensities consistent with the peak fraction. The concentration of proteins present in gradient fractions should be sufficient for direct detection by e.g., silver or Sypro Ruby staining. Protein identities can be confirmed by Western blotting using specific antibodies (see Notes section) and/or mass spectrometry.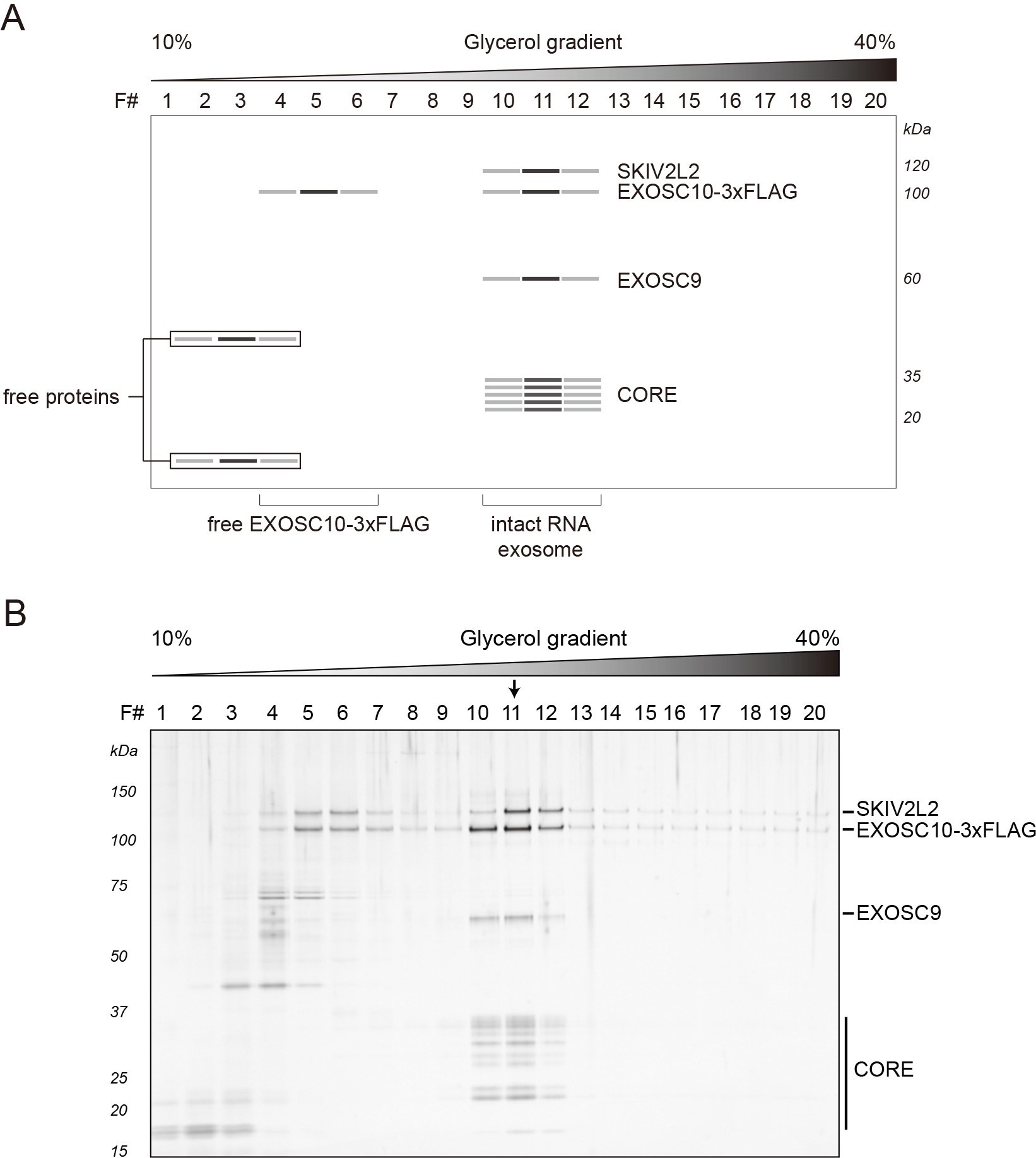
Figure 1. Representative results: RNA exosomes purified from HEK-293 cells expressing EXOSC10-3xFLAG. A. Schematic diagram of a stained SDS-polyacrylamide gel demonstrating protein bands consistent with the separation of EXOSC10-3xFLAG purified RNA exosomes obtained after sedimentation within a 10-40% v/v glycerol gradient. B. The original gel, reproduced from Domanski et al., 2016. Separated proteins were visualized by silver staining. The arrow indicates the peak fraction.
Notes
Listed are some commercially available antibodies that we have used with success to identify exosome components by Western blotting.
- Anti-EXOSC10 antibody (Abcam, catalog number: ab95028)
- Anti-SKIV2L2 antibody (Abcam, catalog number: ab70552)
- Anti-EXOSC3 antibody (Proteintech, catalog number: 15062-1-AP)
Recipes
- ExoI extraction solution
20 mM HEPES-NaOH, pH 7.4
300 mM NaCl
1% Triton X-100 (v/v)
1x protease inhibitor cocktail - ExoII extraction solution
20 mM HEPES-NaOH, pH 7.4
100 mM NaCl
5 mM CHAPS
1x protease inhibitor cocktail - 1 M Tris-HCl, pH 8.0 (for quenching DTSSP crosslinking reactions)
- Gradient solution – ‘light’
10% v/v glycerol
20 mM HEPES-NaOH, pH 7.4
100 mM NaCl
Sterile filter with a 0.45 μm syringe filter - Gradient solution – ‘heavy’
40% glycerol
20 mM HEPES-NaOH, pH 7.4
100 mM NaCl
Sterile filter with a 0.45 μm syringe filter
Note: Many of the solutions used in the NuPAGE® system can be made in the laboratory and do not need to be purchased. Consult the recipes provided by the manufacturer (Life Technologies Corporation). In lieu of this, traditional discontinuous Tris-glycine SDS-PAGE can be carried out using standard methods (Rosenberg, 2005) with comparable results.
Acknowledgments
We thank Professors Michael P. Rout and Torben Heick Jensen for their invaluable support of our research. We also thank Ms. Hua Jiang and Ms. Leila Saba for copyediting. This work was supported in part by the National Institutes of Health grants P41GM109824 and P50GM107632, the Lundbeck Foundation, and the Danish National Research Foundation.
References
- Ahuja, D., Saenz-Robles, M. T. and Pipas, J. M. (2005). SV40 large T antigen targets multiple cellular pathways to elicit cellular transformation. Oncogene 24(52): 7729-7745.
- Cristea, I. M. and Chait, B. T. (2011). Conjugation of magnetic beads for immunopurification of protein complexes. Cold Spring Harb Protoc 2011(5): pdb prot5610.
- Dai, L., Taylor, M. S., O’Donnell, K. A. and Boeke, J. D. (2012). Poly(A) binding protein C1 is essential for efficient L1 retrotransposition and affects L1 RNP formation. Mol Cell Biol 32(21): 4323-4336.
- Domanski, M., Molloy, K., Jiang, H., Chait, B. T., Rout, M. P., Jensen, T. H. and LaCava, J. (2012). Improved methodology for the affinity isolation of human protein complexes expressed at near endogenous levels. Biotechniques 0(0): 1-6.
- Domanski, M., Upla, P., Rice, W. J., Molloy, K. R., Ketaren, N. E., Stokes, D. L., Jensen, T. H., Rout, M. P. and LaCava, J. (2016). Purification and analysis of endogenous human RNA exosome complexes. RNA 22(9): 1467-1475.
- Freshney, R. I. (2011). Culture of Animal Cells. 6th edition. John Wiley & Sons.
- Hakhverdyan, Z., Domanski, M., Hough, L. E., Oroskar, A. A., Oroskar, A. R., Keegan, S., Dilworth, D. J., Molloy, K. R., Sherman, V., Aitchison, J. D., Fenyo, D., Chait, B. T., Jensen, T. H., Rout, M. P. and LaCava, J. (2015). Rapid, optimized interactomic screening. Nat Methods 12(6): 553-560.
- Kilchert, C., Wittmann, S. and Vasiljeva, L. (2016). The regulation and functions of the nuclear RNA exosome complex. Nat Rev Mol Cell Biol 17(4): 227-239.
- LaCava, J., Jiang, H. and Rout, M. P. (2016). Protein complex affinity capture from cryomilled mammalian cells. J Vis Exp(118).
- LaCava, J., Molloy, K. R., Taylor, M. S., Domanski, M., Chait, B. T. and Rout, M. P. (2015). Affinity proteomics to study endogenous protein complexes: pointers, pitfalls, preferences and perspectives. Biotechniques 58(3): 103-119.
- Life Technologies Corporation. NuPAGE® Technical Guide. 3188 (Ed.). Life Technologies Corporation.
- Muller, N., Girard, P., Hacker, D. L., Jordan, M. and Wurm, F. M. (2005). Orbital shaker technology for the cultivation of mammalian cells in suspension. Biotechnol Bioeng 89(4): 400-406.
- Rosenberg, I. M. (2005). Protein analysis and purification. 2nd edition. Birkhäuser Boston.
- Taylor, M. S., LaCava, J., Dai, L., Mita, P., Burns, K. H., Rout, M. P. and Boeke, J. D. (2016). Characterization of L1-ribonucleoprotein particles. Methods Mol Biol 1400: 311-338.
- Taylor, M. S., LaCava, J., Mita, P., Molloy, K. R., Huang, C. R., Li, D., Adney, E. M., Jiang, H., Burns, K. H., Chait, B. T., Rout, M. P., Boeke, J. D. and Dai, L. (2013). Affinity proteomics reveals human host factors implicated in discrete stages of LINE-1 retrotransposition. Cell 155(5): 1034-1048.
- Uphoff, C. C. and Drexler, H. G. (2013). Basic Cell Culture Protocols. 4th edition. In: Helgason, C. D. and Miller, C. L. (Eds.). Humana Press.
Article Information
Copyright
© 2017 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Domanski, M. and LaCava, J. (2017). Affinity Purification of the RNA Degradation Complex, the Exosome, from HEK-293 Cells. Bio-protocol 7(8): e2238. DOI: 10.21769/BioProtoc.2238.
Category
Biochemistry > Protein > Isolation and purification
Molecular Biology > RNA > RNA degradation
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.