- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Heparan Sulfate Identification and Characterisation: Method I. Heparan Sulfate Identification by NMR Analysis
Published: Vol 7, Iss 7, Apr 5, 2017 DOI: 10.21769/BioProtoc.2196 Views: 8990
Reviewed by: Vivien Jane Coulson-ThomasMasahiro MoritaAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Nov 2016
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
Heparin and heparan sulfate (HS) may be purified from complex biological matrices and are often isolated in sub-milligram quantities but not unequivocally identified by spectroscopic means. This protocol details a methodology to rapidly assess the gross compositional features and approximate purity of HS by 1H nuclear magnetic resonance. A complimentary method for identification and characterisation of heparan sulfate can be found at Carnachan and Hinkley (2017).
Keywords: Heparan sulfateBackground
A number of methods exist for the analysis of heparin and HS. This protocol aims to provide a reproducible and widely applicable method for the rapid identification of heparin/HS by nuclear magnetic resonance (NMR). Small samples (~0.3 mg) can be readily assessed in a non-destructive manner to ascertain an approximate purity that identifies other common contaminants found in biological samples when purifying heparin-like molecules. The procedures described herein are intended to provide a stepwise protocol suitable for a laboratory inexperienced in glycosaminoglycan (GAG) analysis.
Materials and Reagents
- Microcentrifuge tubes (1.5 ml) (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 3456 )
- 5 mm NMR tubes (Norell, catalog number: XR-55TM-7 )
- Chondroitin sulfate A (from bovine trachea) (Sigma-Aldrich, catalog number: C8529 )
- Dermatan sulfate (Sigma-Aldrich, catalog number: C3788 )
- Chondroitin sulfate C (from shark cartilage, contains ~10% chondroitin sulfate A) (Sigma-Aldrich, catalog number: C4384 )
- Heparan sulfate (from porcine mucosa) (Celsus Laboratories, catalog number: HO-03103 )
- Heparin (from porcine mucosa) (New Zealand Pharmaceuticals, batch number: 5108356 )
- Heparanoid (from porcine mucosa) (New Zealand Pharmaceuticals, batch number: BX-090-010 )
- Deuterium oxide (99.9 atom%) (Cambridge Isotope Laboratories, catalog number: DLM-4-100 )
- Tert-butanol (t-BuOH, ACS reagent) (Sigma-Aldrich, catalog number: 360538 )
Equipment
- NMR spectrometer (Bruker, model: Bruker Avance DPX-500 )
- Freeze drier (Eyela, model: FD-1 )
- Pipettors (0.5-10 μl, 20-200 μl and 100-1,000 μl) (Eppendorf)
Software
- Software Topspin 2.6
Procedure
- Sample preparation
Exchangable protons in the sample are treated to a deuterium exchange regime before NMR analysis:- Add D2O (1 ml) to a sample (~12 mg) of the glucosaminoglycan in an Eppendorf tube and agitate the sample to complete dissolution.
- The sample is freeze dried in the ‘loosely capped’ tube and this process repeated twice more.
- Finally the sample is made up with D2O (0.7 ml) containing 0.2 mg/ml t-BuOH as an internal reference (δ 1H 1.24 ppm) and transferred to the NMR tube for analysis.
- Add D2O (1 ml) to a sample (~12 mg) of the glucosaminoglycan in an Eppendorf tube and agitate the sample to complete dissolution.
- Sample concentration
The concentration of 17 mg/ml has been found to be the minimum necessary for highly reproducible 2-dimensional NMR. The 1H NMR spectra of this class of compound is significantly affected by pH, cross-linking capable species (e.g., Ca2+) and the salt form of the sulfated moieties (Liu et al., 2009; Guerrini et al., 2008). If such contamination is suspected, the sample should be converted to the sodium salt and dialysed (3,500 MW cutoff) against water exhaustively, before freeze-drying and deuterium exchange. - Data collection
- Spectra were collected at 30 °C on a three-channel Bruker Avance III 500 (installed August 2007). Proton frequency was 499.843 MHz and the probe was a Bruker two channel 5-mm broadband observe nuclei probe (31P-109Ag) equipped with actively shielded 50-G/cm Z-axis Pulsed Field Gradients (PFG).
- Data acquisition used a spectral width of 10,302 Hz, 65,536 data points, 30-degree excitation pulse, 32 transients, each with a 2-sec delay time and an FID acquisition time of 1.59 sec.
- Spectra with solvent-suppression were recorded: low-power continuous-wave pre-saturation was followed by a composite-observe pulse utilizing the ‘zgcppr’ Bruker sequence (software Topspin 2.6).
Data analysis
- The individual GAG’s are readily identified by their distinctive 1H NMR spectra (Figure 1). The presence of contaminating chondroitin or dermatan sulfate in a sample of heparan sulfate or heparin is apparent by examination of the glycosaminoglycan acetate resonances (Carnachan, 2016). For both heparin and heparan sulfate this acetate resonance occurs at δ 2.034 ppm and any additional acetate resonance downfield indicates the presence of other GAG’s (see Figure 2). In addition, heparin and heparan sulfate have resonances at δ 3.25, 3.37 and > 5.0 ppm that distinguish them from chondroitins or dermatan.
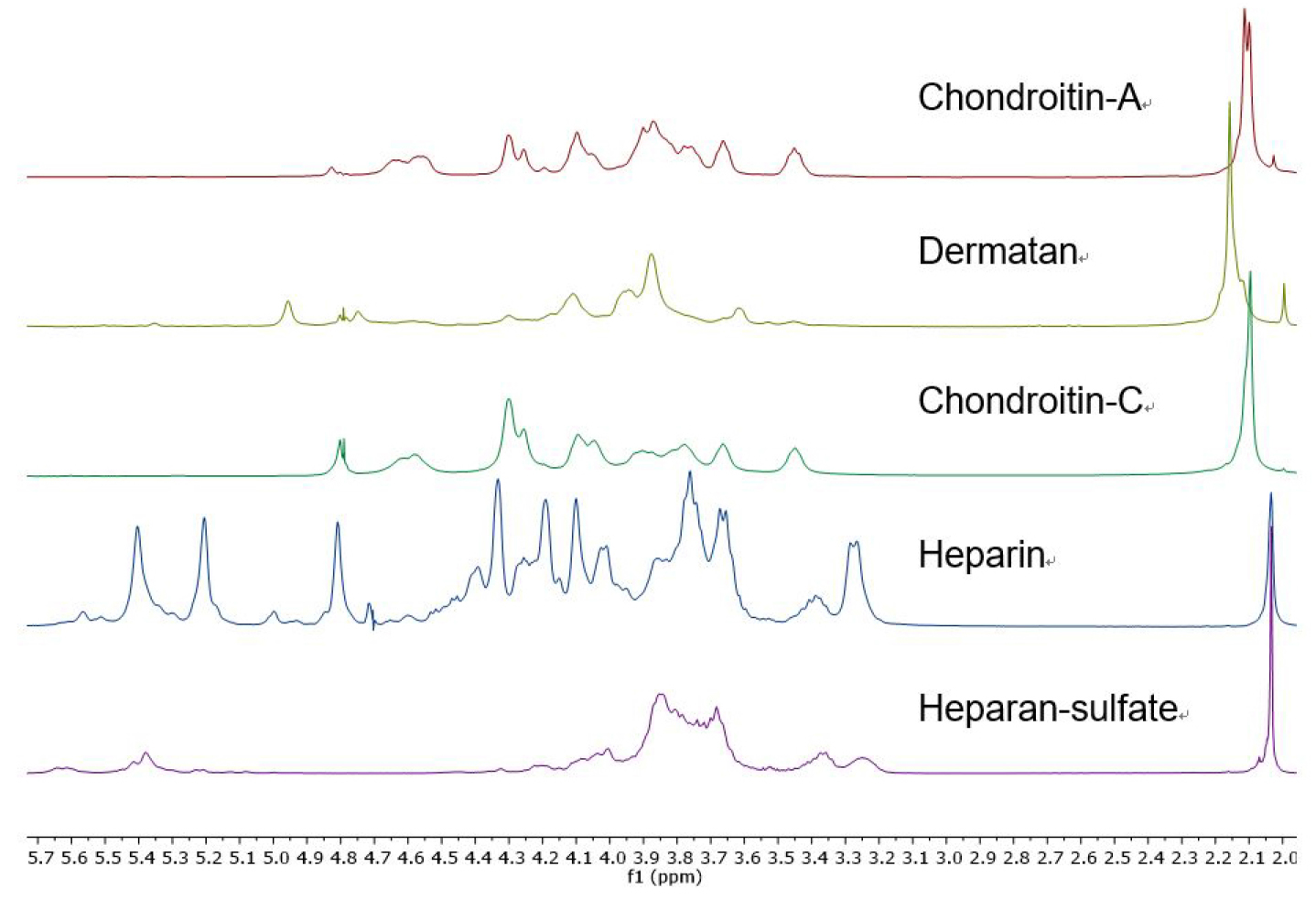
Figure 1. 1H NMR spectra (D2O, 25 °C, with HDO suppression, referenced to t-BuOH) of heparin, heparan sulfate, chondroitin sulfates and dermatan sulfate. Chondroitin-A is a mixture of chondroitins A&C.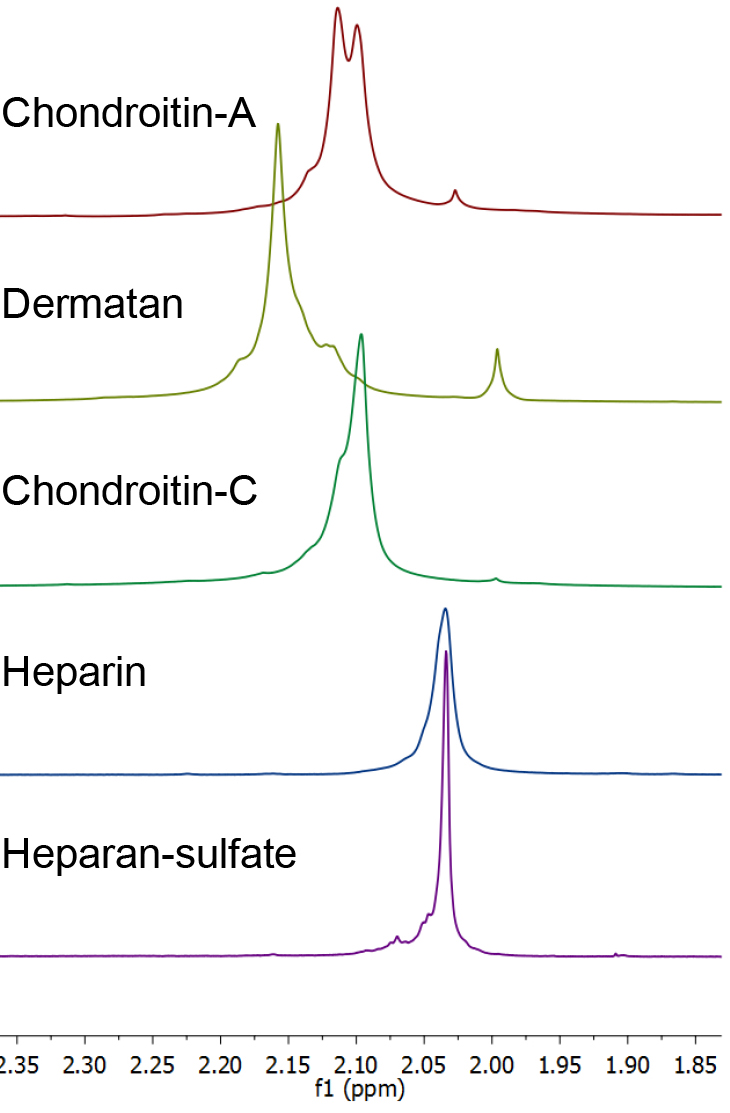
Figure 2. 1H NMR spectra of the acetate methyl resonances (D2O, 25 °C, with HDO suppression, referenced to t-BuOH) of heparin, heparan sulfate, chondroitin sulfates and dermatan sulfate. Chondroitin-A is a mixture of chondroitins A&C. - By way of demonstration, 1H NMR spectra that are typical of those recorded during a purification process from Heparanoid (see Note 6) are presented in Figure 3.
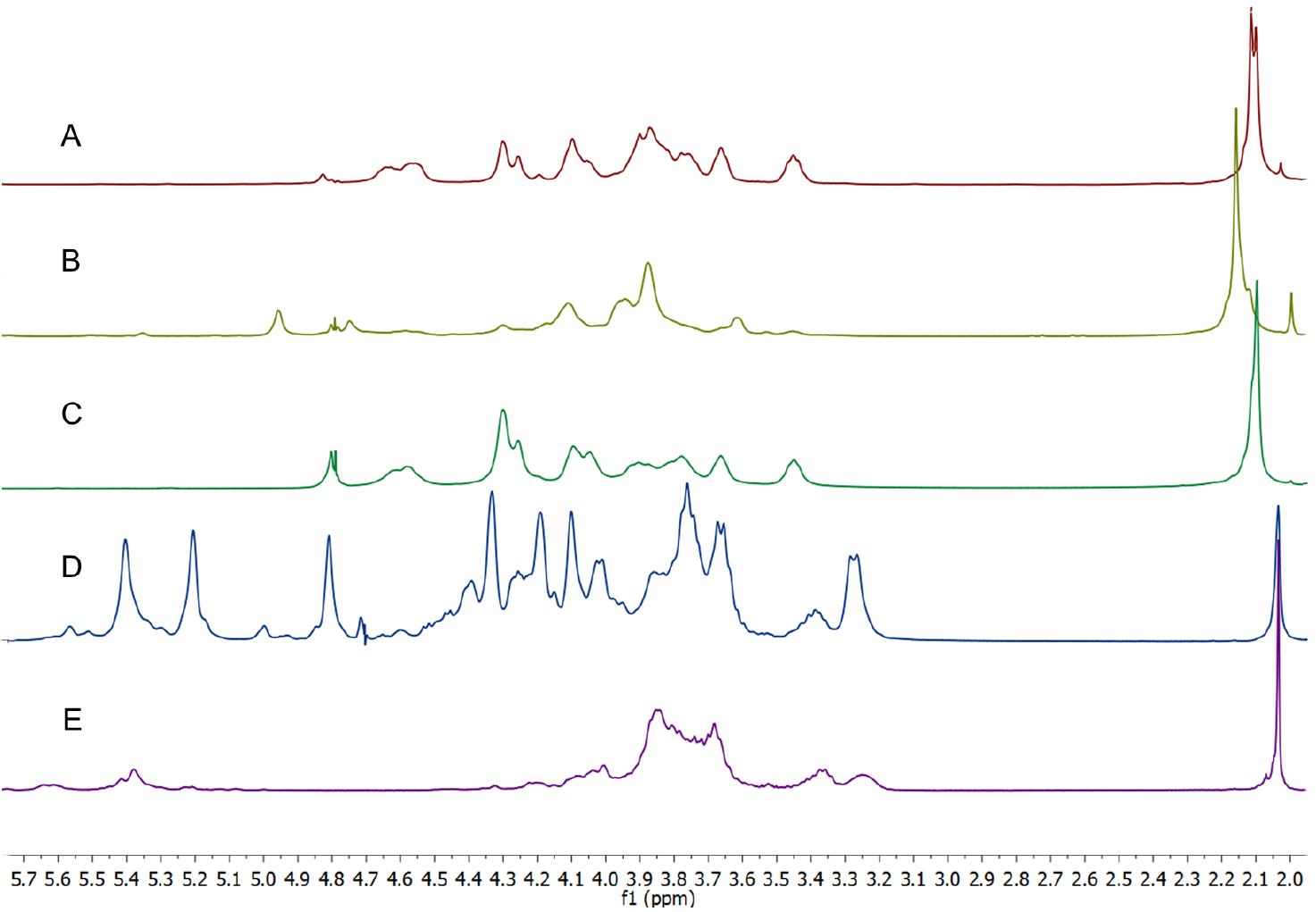
Figure 3. 1H NMR spectra of Heparanoid and partially purified fractions obtained from DEAE-Sepharose chromatographic separation. A. Very crude Heparanoid with significant protein contribution; B. Heparanoid - different batch to A; C and D. GAG mixtures that have a significant proportion of heparin-like materials; E. Fraction that is > 85% HS (D2O, 25 °C, with HDO suppression [discontinuous peak at δ 4.6 ppm], referenced to t-BuOH). - The co-occurrence of the heparin and heparan sulfate N-acetate methyl resonances at δ 2.034 ppm make using this peak for assessing the relative proportion of these species in a sample problematic. However, the intensity of this peak relative to the remainder of the spectrum can be indicative of whether a sample is more heparan sulfate-like, or, more heparin-like. The peak height for the acetate resonance of heparin is equivalent to the peak height of the methine resonances observed at δ ~3.8 ppm, while for heparan sulfate the acetate resonance is ~2.5 fold greater than the methine resonances (see Figure 2). Low levels (even up to < 15%) of contaminating heparin in a sample of heparan sulfate are very difficult to confirm or quantitate by 1H NMR (Keire et al., 2010), and are more readily assessed using 2-D NMR techniques (Guerrini et al., 2005).
- A semi-quantitative purity assessment can be undertaken for samples containing primarily one GAG (> 90%). Using a known concentration of t-BuOH in D2O and a pure sample of a particular GAG a standard curve may be generated. GAG was weighed out accurately into ~10, 5, 2 and 0.5 mg aliquots and made up with exactly 1.0 ml of t-BuOH standard solution for NMR analysis. Using integrated areas, the ratio of the acetate peak of the GAG divided by the t-BuOH resonance plotted against the known amount of GAG in solution generates a straight-line relation (Figure 4). A sample of unknown purity made up in the same concentration and volume of t-BuOH solution can therefore be assessed for purity compared to the reference material. Such a standard curve is particularly useful in assessing possible contributions from salt and water concentrations that are effectively invisible to this NMR analysis and not trivial to measure in a non-destructive manner on small samples (< 5 mg).

Figure 4. Standard curve plotting the ratio of acetate and internal-standard resonances (integrated areas from 1H NMR) against the known mass of HS (> 95% w/w) in the sample (t-BuOH standard solution 0.20 mg/ml)
Notes
- GAG’s, either as a dry lyophilized solid or as a dilute solution (D2O, 5 °C) are stable indefinitely (> 5 yr). Dry solids appear to undergo no degradation over time either at room temperature (RT) or freezer conditions (to -70 °C).
- Completing the D2O exchange process has only a subtle effect on the NMR spectra, and so for routine analysis or during purification-monitoring this step is often omitted.
- Concentrated samples (> 60 mg/ml) (prepared for example for 13C analysis or rapid 2-D NMR techniques) exhibit significant line broadening and chemical shift changes in the 1H NMR spectrum, thus reference 1H NMR must be recorded as dilute solutions only.
- We have demonstrated in the laboratory that freshly lyophilized HS will adsorb 13% w/w of water on standing. This is a reproducible mass gain on standing and readily-reversible process by lyophilization. Samples are routinely stored at -70 °C and so are allowed to warm to RT in air-tight containers before exposure to the atmosphere to minimize water uptake during handling.
- The presence of salts can be subjectively noted by the physical appearance of the GAG. Heparin and HS are fluffy white ‘cotton-wool’ like solids. The presence of salt at ~> 10% w/w generates a more rigid, tough off-white solid which adsorbs atmospheric water at an accelerated rate compared to the de-salted material. This is particularly important to note for weighing small amounts of HS that are subsequently entering a dose-response bioassay.
- Heparanoid is a complex mixture of GAG’s recovered from the side-streams produced in the production of Heparin from porcine mucosa. The composition of material described as Heparanoid varies significantly (see Figures 3i and 3ii), dependant on which side-streams of the Heparin process are combined. Thus the levels of protein, individual GAG’s (including Heparin), salts and other biological materials can vary wildly in individually sourced materials and discrete batches. One method (Shworak, 2001) of purifying HS is partitioning on a diethylaminoethyl-Sepharose (DEAE-Sepharose) column. Column elution may be achieved by step-wise increments in ionic strength (e.g., sodium chloride in phosphate buffer). For analysis by NMR fractions are desalted (dialysis), lyophilized and the resultant amorphous white solid exchanged with D2O before 1H NMR spectra recorded (unpublished work).
Acknowledgments
This research was supported in part by the New Zealand Ministry of Business, Innovation and Employment, and the Kiwi Innovation Network (KiwiNet, VL001298). The collaborative research completed with Drs Simon M. Cool, Victor Nurcombe and R. Alex A. Smith (Institute of Medical Biology, Agency for Science, Technology and Research, Immunos, Singapore) is acknowledged.
References
- Carnachan, S. M., Bell, T. J., Sims, I. M., Smith, R. A., Nurcombe, V., Cool, S. M. and Hinkley, S. F. (2016). Determining the extent of heparan sulfate depolymerisation following heparin lyase treatment. Carbohydr Polym 152: 592-597.
- Carnachan, S. C. and Hinkley, S. F. R. (2017). Heparan sulfate identification and characterisation: Method II. Enzymatic depolymerisation and SAX-HPLC analysis to determine disaccharide composition. Bio-protocl 7(07): e2197.
- Guerrini, M., Beccati, D., Shriver, Z., Naggi, A., Viswanathan, K., Bisio, A., Capila, I., Lansing, J. C., Guglieri, S., Fraser, B., Al-Hakim, A., Gunay, N. S., Zhang, Z., Robinson, L., Buhse, L., Nasr, M., Woodcock, J., Langer, R., Venkataraman, G., Linhardt, R. J., Casu, B., Torri, G. and Sasisekharan, R. (2008). Oversulfated chondroitin sulfate is a contaminant in heparin associated with adverse clinical events. Nat Biotechnol 26 (6): 669-675.
- Guerrini, M., Naggi, A., Guglieri, S., Santarsiero, R. and Torri, G. (2005). Complex glycosaminoglycans: profiling substitution patterns by two-dimensional nuclear magnetic resonance spectroscopy. Anal Biochem 337 (1): 3547.
- Keire, D. A., Mans, D. J., Ye, H., Kolinski, R. E. and Buhse, L. F. (2010). Assay of possible economically motivated additives or native impurities levels in heparin by 1H NMR, SAX-HPLC, and anticoagulation time approaches. J Pharm Biomed Anal 52(5): 656-664.
- Liu, H., Zhang, Z. and Linhardt, R. J. (2009). Lessons learned from the contamination of heparin. Nat Prod Rep 26 (3): 313-321.
- Shworak, N. W. (2001). High-specific-activity 35S-labeled heparan sulfate prepared from cultured cells. In: Iozzo, R. V. (Ed.). Proteoglycan Protocols. Methods in Molecular Biology pp: 79-89.
Article Information
Copyright
© 2017 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Carnachan, S. M. and Hinkley, S. F. R. (2017). Heparan Sulfate Identification and Characterisation: Method I. Heparan Sulfate Identification by NMR Analysis. Bio-protocol 7(7): e2196. DOI: 10.21769/BioProtoc.2196.
Category
Biochemistry > Carbohydrate > Disaccharide
Biochemistry > Carbohydrate > Polysaccharide
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.