- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Heterochronic Pellet Assay to Test Cell-cell Communication in the Mouse Retina
Published: Vol 7, Iss 3, Feb 5, 2017 DOI: 10.21769/BioProtoc.2117 Views: 11022
Reviewed by: Oneil G. BhalalaAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Sep 2016

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
All seven retinal cell types that make up the mature retina are generated from a common, multipotent pool of retinal progenitor cells (RPCs) (Wallace, 2011). One way that RPCs know when sufficient numbers of particular cell-types have been generated is through negative feedback signals, which are emitted by differentiated cells and must reach threshold levels to block additional differentiation of that cell type. A key assay to assess whether negative feedback signals are emitted by differentiated cells is a heterochronic pellet assay in which early stage RPCs are dissociated and labeled with BrdU, then mixed with a 20-fold excess of dissociated differentiated cells. The combined cells are then re-aggregated and cultured as a pellet on a membrane for 7-10 days in vitro. During this time frame, RPCs will differentiate, and the fate of the BrdU+ RPCs can be assessed using cell type-specific markers. Investigators who developed this pellet assay initially demonstrated that neonatal RPCs give rise to rods on an accelerated schedule compared to embryonic RPCs when the two cell types are mixed together (Watanabe and Raff, 1990; Watanabe et al., 1997). We have used this assay to demonstrate that sonic hedgehog (Shh), which we found acts as a negative regulator of retinal ganglion cell (RGC) differentiation, promotes RPC proliferation (Jensen and Wallace, 1997; Ringuette et al., 2014). More recently we modified the heterochronic pellet assay to assess the role of feedback signals for retinal amacrine cells, identifying transforming growth factor β2 (Tgfβ2) as a negative feedback signal, and Pten as a modulator of the Tgfβ2 response (Ma et al., 2007; Tachibana et al., 2016). This assay can be adapted to other lineages and tissues to assess cell-cell interactions between two different cell-types (heterotypic) in either an isochronic or heterochronic manner.
Keywords: Heterochronic pellet assayBackground
Several mechanisms are employed to ensure that the correct numbers of differentiated cells are generated during organ and tissue development. For example, progenitor cells may respond to the levels of hormones or growth factors secreted by differentiated cells, progenitors may count the number of divisions they undergo, or there may be a mechanism to count the final number of differentiated cells (Lui and Baron, 2011). In the retina, negative feedback signals that are secreted by differentiated cells are sensed by progenitor cells, which stop producing that differentiated cell type when the signals reach threshold levels (Belliveau and Cepko, 1999; Reh and Tully, 1986; Waid and McLoon, 1998). We and other have demonstrated that Shh is an essential negative regulator of a RGC fate (Wang et al., 2005; Zhang and Yang, 2001). We also dissected the feedback process for retinal amacrine cells, showing that the transcription factor Zac1 acts in amacrine cells to initiate transforming growth factor b2 (Tgfb2) expression, which negatively regulates RPC proliferation and amacrine cell differentiation (Ma et al., 2007). Notably, other TGFβ family members have similar feedback functions in the olfactory epithelium (Wu et al., 2003), pancreas (Harmon et al., 2004), and skeletal muscle (Tobin and Celeste, 2005). We also used the heterochronic pellet assay to examine how amacrine cell feedback signals are themselves regulated. We found that Pten is an essential positive regulator of amacrine cell differentiation, and using the pellet assay, we demonstrated that Pten acts in RPCs to control responsiveness to Tgfβ2 signaling (Tachibana et al., 2016). Understanding how amacrine cells and RPCs interact provides important new insights into how cell number is controlled in the retina. Notably, similar interactions between Pten and Tgfβ signaling may underlie cell number control in other vertebrate organs where Tgfβ signaling is an important determinant of organ size.
Materials and Reagents
- Fisherbrand sterile 100 x 15 mm polystyrene Petri dish (Thermo Fisher Scientific, Fisher Scientific, catalog number: FB0875713 )
- 15 ml conical tubes (SARSTEDT, catalog number: 62.554.502 )
- Sterile individually packaged 5 ml pipettes (SARSTEDT, catalog number: 86.1253.001 )
- Tissue culture 24-well plates (SARSTEDT, catalog number: 83.3922.300 )
- SamcoTM extra long transfer pipet (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 262 )
- 13 mm, 0.8 μm Nuclepore Track-Etch membrane (GE Healthcare, catalog number: 110409 )
- Kimwipes (Kimberly-Clark Worldwide, catalog number: 34120 ) (not autoclaved, but kept clean)
- Superfrost Plus Micro slides (VWR, catalog number: 48311-703 )
- Standard microscope slide box (Heathrow Scientific, catalog number: HEA15991A )
- Micro cover glass (VWR, catalog number: 48404-454 )
- 4 in x 250 ft Parafilm roll (Bemis, catalog number: PM999 or VWR, catalog number: 52858-032 )
- 50 ml corning tube (SARSTEDT, catalog number: 62.547.254 )
- 0.22 μm sterilize filter filtropur (SARSTEDT, catalog number: 83.1826.001 )
- 60 ml syringe (Medtronic, catalog number: 8881560125 )
- Rapid-FlowTM sterile disposable bottle top filters (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 291-4520 )
- E15.5 CD1 mouse (Charles River Laboratories International, catalog number: 022 ) (see Note 1)
- P2 Ptenfl/fl; Pax6-Cre+ or Pten+/+ mouse (see Note 2)
- 1x Ca2+/Mg2+-free DPBS (Thermo Fisher Scientific, GibcoTM, catalog number: 14190250 )
- Trypan blue (Thermo Fisher Scientific, GibcoTM, catalog number: 15250061 )
- Aqua-Poly/Mount (Polysciences, catalog number: 18606 )
- Trypsin (Sigma-Aldrich, catalog number: T1005 )
- Heat inactivated FBS (fetal bovine serum) (Thermo Fisher Scientific, GibcoTM, catalog number: 12484028 )
- DMEM (Thermo Fisher Scientific, GibcoTM, catalog number: 11965092 )
- 1x HBSS (Thermo Fisher Scientific, GibcoTM, catalog number: 24020117 )
- Heat inactivated horse serum (Thermo Fisher Scientific, GibcoTM, catalog number: 26050088 )
- 200 mM L-glutamine (Sigma-Aldrich, catalog number: G7513 )
- HEPES (Thermo Fisher Scientific, GibcoTM, catalog number: 15630080 )
- Penicillin-streptomycin (Thermo Fisher Scientific, GibcoTM, catalog number: 15140122 )
- Amphotericin B (Thermo Fisher Scientific, GibcoTM, catalog number: 15290026 )
- 5-Bromouridine (BrdU) (Sigma-Aldrich, catalog number: 850187 )
- Sodium chloride (NaCl) (Sigma-Aldrich, catalog number: S3014 )
- Potassium chloride (KCl) (EMD Millipore, catalog number: PX1405 )
- Potassium phosphate monobasic (KH2PO4) (Sigma-Aldrich, catalog number: P5379 )
- Sodium phosphate dibasic (Na2HPO4) (Sigma-Aldrich, catalog number: S7907 )
- Crystalline PFA (Sigma-Aldrich, catalog number: P6148 )
- Sucrose (Sigma-Aldrich, catalog number: S9378 )
- 11.8 M hydrochloric acid (HCl) (Sigma-Aldrich, catalog number: 258148 )
- Triton X-100 (Sigma-Aldrich, catalog number: T8787 )
- 4’,6-diamidino-2-phenylindole, dihydrochloride (DAPI) as pre-prepared working solution that is used as described by the manufacturer (Thermo Fisher Scientific, Molecular ProbesTM, catalog number: D1306 )
- Antibodies against Pax6 and BrdU (see Recipes, Table 1)
Pax6 (BioLegend, catalog number: 901301 )
BrdU (Bio-Rad Laboratories, catalog number: OBT0030 )
Donkey Anti-rabbit Alexa-Fluor 488 (Thermo Fisher Scientific, InvitrogenTM, catalog number: A-21206 )
Goat Anti-rat Alexa Fluor 568 (Thermo Fisher Scientific, InvitrogenTM, catalog number: A-11077 ) - Optimal cutting temperature compound (VWR, catalog number: 95057-838 )
- 2.5% trypsin (see Recipes)
- 0.125% trypsin (see Recipes)
- Retinal explant media (REM) (see Recipes)
- 1 mg/ml BrdU (see Recipes)
- 10x phosphate-buffered saline (PBS) (see Recipes)
- 1x phosphate-buffered saline (PBS) (See Recipes)
- 20% paraformaldehyde (PFA) (see Recipes)
- 4% paraformaldehyde (PFA) (See Recipes)
- 20% sucrose (see Recipes)
- 2 N hydrochloric acid (see Recipes)
- 1x phosphate-buffered saline/0.1% Triton X-100 (PBT) (see Recipes)
- Blocking solution (see Recipes)
Equipment
- Dumont forceps #5 (Fine Science Tools, catalog number: 11252-20 )
- Dumont forceps #55 (Fine Science Tools, catalog number: 11255-20 )
- Dumont forceps AA (Fine Science Tools, catalog number: 11210-20 )
- Shallow form shaking water bath (Precision Scientific, catalog number: 66799 )
- Pipette pump (SP Scienceware - Bell-Art Products - H-B Instrument, catalog number: F37898-0000 )
- 37 °C, 5% CO2 water jacketed incubator (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 3110 )
- Refrigerated tabletop centrifuge for 15 ml conical tubes (Eppendorf, model: 5810 R )
- Hemacytometer chamber (Hausser Scientific, catalog number: 3100 )
- P20 pipetmen (Gilson, catalog number: F123600 )
- P200 pipetmen (Gilson, catalog number: F123601 )
- P1000 pipetmen (Gilson, catalog number: F123602 )
- Cryostat (Leica Biosystems, model: CM3050 S )
- -20 °C freezer
- Upright fluorescence microscope (Leica Microsystems, model: DM RXA2 )
- Autoclave
- 1 L beaker (Corning, Pyrex®, catalog number: 1395-1L )
- 500 ml beaker (Corning, Pyrex®, catalog number: 1395-500 )
- 250 ml Erlenmeyer flask (Corning, Pyrex®, catalog number: 4450-250 )
- Fume hood
- Stereomicroscope for dissection (Leica Microsystems, model: MZ6 )
- Inverted light microscope (Leica Microsystems, model: DMIL LED )
Software
- GraphPad Prism (GraphPad Software)
Procedure
Notes:
- Pre-warm 0.125% trypsin and REM (see Recipes) to 37 °C prior to the experiment.
- This protocol was performed without sterile technique, but we took extreme caution to keep the cells and equipment as clean as possible.
- Dissociating E15.5 mouse retina
- Enucleate the eyes from E15.5 CD1 embryos at room temperature (see Note 1 about mice).
- Place eyes in Petri dish and add cold 1x DPBS to cover (Figure 1A).
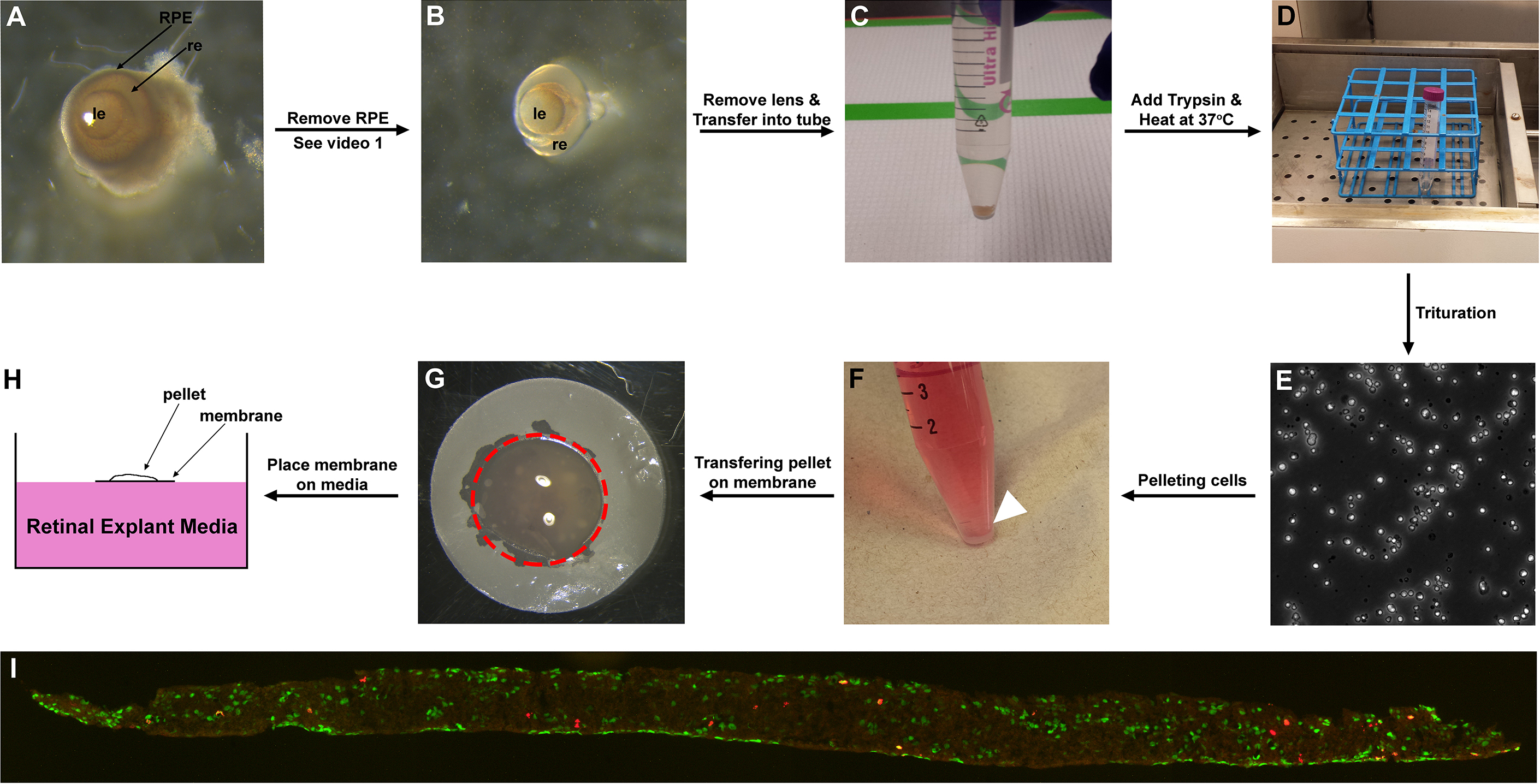
Figure 1. Representative images of retina/retinal cells at different time points of the protocol. A. E15.5 eyes after enucleation; B. E15.5 retina after RPE removal; C. E15.5 retina sunk to the bottom of a 15 ml Corning tube; D. Trypsinizing the retinas in 37 °C water bath for 10 min; E. Dissociated retinal cells. F. Heterochronic retinal cell aggregate pelleted on the bottom of the 15 ml Corning tube (arrowhead); G. Cell pellet placed on the membrane (outlined by red dotted line); H. Cell pellet on membrane placed on the surface of REM (side view); I. Section of aggregate labelled with BrdU (red) and amacrine cell marker Pax6 (green). le: lens, re: retina, RPE: retinal pigment epithelium. - Remove retinal pigment epithelium (RPE) and lens from eyes using Dumont forceps #55 and #5 (Figure 1B) (see Note 4). To remove RPE, grab a small area of the sclera with Dumont forceps #55, then stab onto the sclera with Dumont forceps #5 to cause incision. Grind the edge of the incision with both forceps, and slowly rip open the incision. Keep opening up the incision until sclera, cornea and optic nerve are removed. RPE should detach from retina along with cornea (Video 1). Keeping the eyes in cold 1x DPBS aids RPE detachment. Detach ciliary margin from the retina using the forceps. This loosens the lens attachment on the retina. Gently pull the lens out (Video 2). Replace 1x DPBS for every eye dissection to keep Petri dish and buffer cold.
 Video 1. Removing RPE from eye Video 2. Removing lens from eye
Video 1. Removing RPE from eye Video 2. Removing lens from eye - Transfer retinas into 15 ml Corning tube (with 10 ml of 1x DPBS at room temperature) (Figure 1C). Allow retinas to sink to bottom of the tube. (We recommend collecting ~20 retinas [2 eyes from each of 10 embryos] for this protocol. This will give approximately 8.0 x 106 cells.)
- Remove as much of the 1x DPBS as possible using a 5 ml pipet. Make sure not to pipet out the retinal tissue.
- Add 100 μl of 0.125% trypsin (pre-warmed to 37 °C) to retinas, and incubate in 37 °C water bath for 10 min (Figure 1D). Please note that retina will not break apart on its own. Trituration is required to break cell-cell interactions.
- Add 5 ml of 20% FBS/1x DPBS to stop trypsinization.
- Homogenize retinal cells completely by pipetting up and down several times (i.e., at least 30 times) using a 5 ml pipet (i.e., triturate; Figure 1E) until no clusters of cells are observed macroscopically. Make sure to triturate gently to avoid any air bubbles as bubbles can damage cells. We recommend using a Pipette pump to make sure trituration is gentle. (Video 3) (see Note 5) Video 3. Triturating trypsinized retinal cells
- Spin down cells at 520 x g for 5 min, then remove as much solution as possible.
- Re-suspend cells in 1 ml of REM (pre-warmed to 37 °C).
- Add 3 µl of 1 mg/ml BrdU into cell resuspension. Vortex gently and briefly to mix.
- Incubate cell resuspension for 1 h at 37 °C, 5% CO2 in water jacketed incubator.
- Enucleate the eyes from E15.5 CD1 embryos at room temperature (see Note 1 about mice).
- Dissociating P2 mouse retina
- Meanwhile, dissect P2 mouse retinas by enucleating the eyes, and removing RPE and lens as described above (see Note 2 and Note 3 about mice).
- Transfer retinas into 15 ml Corning tube (with 10 ml of 1x DPBS at room temperature), keeping the two retinas from each mouse together in a separate tube as the genotype of each mouse is different. Allow retinas to sink to tube bottoms.
- Remove as much 1x DPBS as possible, then add 100 µl of 0.125% trypsin, and incubate in 37 °C water bath for 10 min.
- Stop the reaction by adding 5 ml of 20% FBS/1x DPBS, and triturate the cells by pipetting gently several times with 5 ml pipet and pipette pump.
- Spin down at 520 x g for 5 min, and remove solution as much as possible.
- Re-suspend the cells with 1 ml REM (pre-warmed to 37 °C).
- Meanwhile, dissect P2 mouse retinas by enucleating the eyes, and removing RPE and lens as described above (see Note 2 and Note 3 about mice).
- Preparation of heterochronic cell pellet
- Remove E15.5 CD1 dissociated cells from the incubator after 1 h incubation in BrdU.
- Spin down cells at 520 x g for 5 min, remove media as much as possible, then re-suspend in 1 ml REM. Repeat this step twice more to completely remove any trace of BrdU. Re-suspend the cells in 1 ml REM (pre-warmed to 37 °C).
- For both E15.5 and P2 cells, take 10 µl of the cell resuspension and mix with 10 µl trypan blue. Calculate the concentration of each cell resuspension using hemacytometer (calculation described in Figure 2). Make sure that the cells are dissociated completely (i.e., no clumps of cells or doublets). If you see any clumps, we recommend to re-dissociate the cells.
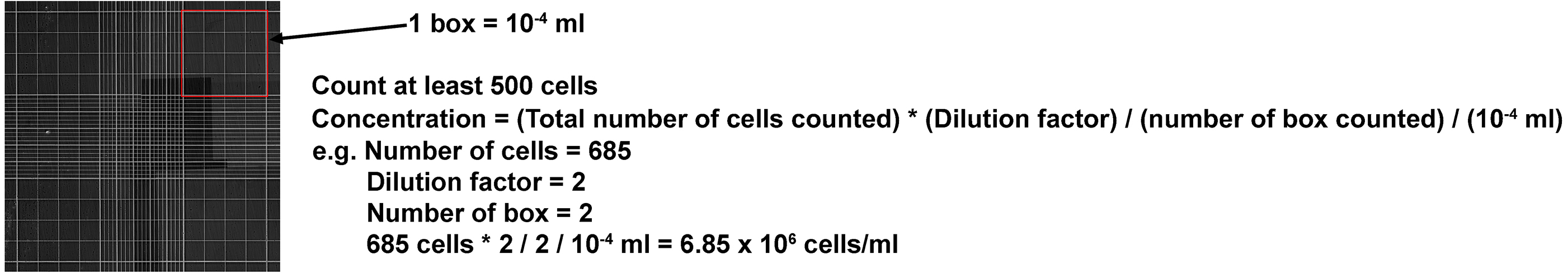
Figure 2. Formula for calculating concentration of cell resuspension using hemacytometer. Note that the dilution factor is 2 as cell resuspension is diluted with trypan blue (10 μl + 10 μl). - Dilute E15.5 cell resuspension to 1 x 106 cells/ml in REM. Aliquot 1 x 106 cells (1 ml) into 15 ml Corning tubes as a control. Re-dilute E15.5 cell resuspension to 0.5 x 106 cells/ml.
- Dilute P2 cell resuspension to 1.0 x 106 cells/ml in REM. Aliquot 1 x 106 cell (1 ml) into 15 ml Corning tubes. Add 100 µl (5.0 x 104 cells) of 0.5 x 106 cells/ml from E15.5 CD1 cell resuspension. Mix thoroughly by pipetting up and down several times with 5 ml pipet with pipette pump.
- Spin down at 520 x g for 5 min, then at 300 x g for 3 min to pellet the heterochronic cell mixtures (Figure 1F).
- Remove E15.5 CD1 dissociated cells from the incubator after 1 h incubation in BrdU.
- Culturing (Video 4)
- Aliquot 1.5 ml REM per well of a 24-well plate.
- Cut the tip of the extra long transfer pipet with scissors, then flame the cut site to smoothen the surface. This will prevent damaging the pellet, and cells adhering to the pipet surface.
- After spinning down the heterochronic cell aggregate (from step C6), dislodge the aggregate from the bottom of the tube by taking some liquid off with a P1000 pipetman. Gently dispense liquid back into the tube at the edge of the pellet in order to dislodge the pellet (see Notes 6 and 7).
- Place dry Nucleopore Track-Etch Membrane on the sterilized Kimwipe sitting on sterile Petri dish lid. Place the tip of the Dumont forceps AA on the edge of the membrane to prevent membrane from rolling when cell pellet is transferred.
- Transfer cell pellet with extra long transfer pipet onto the centre of a dry Nuclepore Track-Etch Membrane (Figure 1G). Note that it is extremely critical to drop the pellet on the centre of the membrane. If pellet is dropped on the edge of the membrane, you could lose the cell pellet as it will flow towards Kimwipe and away from the membrane. Kimwipe should absorb the liquid leaving only the pellet on the membrane.
- Dry the membrane further by placing the membrane on a sterilized Kimwipe. Leave no trace of liquid on the membrane, as it will increase the chance of the membrane sinking in the media, which will result in cell death.
- Grab edge of the membrane with the cell pellet using Dumont forceps AA and gently place it onto the surface of the REM in one well of the 24-well plate (Figure 1H). Make sure the membrane is floating on the media, and does not sink as cells in the pellet will die if submersed.
- Incubate cell aggregate at 37 °C, 5% CO2 for 8 days in vitro (DIV). Replace ½ of the media (750 µl) every second day. Note that it is critical that the membrane is never submerged, so extra care is required when transferring plates in and out of the incubator. Video 4. Transferring heterochronic cell aggregate onto the membrane
- Aliquot 1.5 ml REM per well of a 24-well plate.
- Cell collection and analysis
- Replace REM with 1 ml of 1x DPBS to wash aggregate. Rinse with 1x DPBS two more times to remove trace of REM. Make sure cell pellets are not dislodged from the membrane.
- Remove as much 1x DPBS as possible, then add 500 µl of 4% PFA. Fix aggregate overnight at 4 °C. Note that a shorter fixation time may be required for certain antibodies.
- Wash aggregates three times for 10 min with 1x PBS, then immerse into 20% sucrose/1x PBS for cryoprotection overnight at 4 °C.
- Embed aggregates on membranes into optical cutting temperature compound (OCT) and freeze on dry ice (Figure 3). The membrane should be embedded perpendicular to the bottom of the mold to give a radial section through the retina. We recommend embedding replicate aggregates into the same block.
- Section aggregate on a cryostat at 10 µm, and collect sections on Superfrost Plus slides. We usually collect approximately seven sections per slide on up to 50 slides per block. Place the slides into a Standard microscope slide box. Slides can either be immunostained directly or stored. For storage, tape slide box with masking tape to close completely and prevent moisture from entering. Store box in -20 °C freezer.
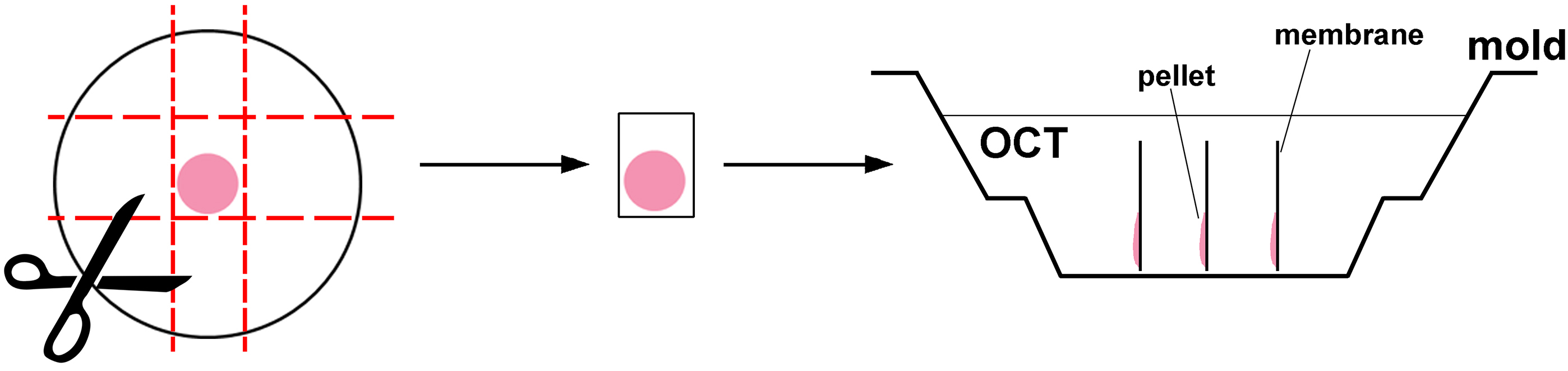
Figure 3. Orientation of heterochronic cell aggregate on the membrane and in the mold when embedding into OCT
- Replace REM with 1 ml of 1x DPBS to wash aggregate. Rinse with 1x DPBS two more times to remove trace of REM. Make sure cell pellets are not dislodged from the membrane.
- Immunostaining
- Bring the slide box to room temperature for 30 min before opening, then remove slides.
- Place slides in pre-warmed 2 N hydrochloric acid and incubate at 37 °C for 20 min.
- Rinse slides in distilled H2O several times to remove as much hydrochloric acid as possible.
- Wash three times for 5 min in 1x PBS to equilibrate the pH of the slides.
- Remove slides from 1x PBS and apply paper towel on the edge of the slide to remove excess liquid.
- Apply 400 μl of blocking solution (10% horse serum/1x PBT) per slide and incubate at room temperature for 1 h. Make sure blocking solution is spread thoroughly to cover the entire surface of the slides.
- Remove as much blocking solution as possible, then apply 200 μl of primary antibodies diluted in blocking solution (see Table 1 for dilution) and place Parafilm to cover entire surface of the slides. Incubate at 4 °C overnight.
- Wash slides three times for 10 min in 1x PBS.
- Apply 200 μl of secondary antibodies (see Table 1) and DAPI diluted in 1x PBT (1:500 and 1:500 dilution respectively) and place Parafilm on top to cover solution. Incubate at room temperature for 1 h. Avoid incubating under light as fluorescence is light-sensitive.
- Wash slides three times for 10 min in 1x PBS.
- Apply Aqua-Poly/Mount and mount with coverslip.
- Take several images (3 or more) of sections of the retinal cell aggregates with upright fluorescence microscope (Figure 1I).
- Bring the slide box to room temperature for 30 min before opening, then remove slides.
Data analysis
- For experimental design, to compare two data sets, we perform the pellet assay a minimum of three times. To compare three or more data sets, we perform a minimum of four experiments. For each experiment, we use a minimum of three replicates. For each replicate, we analyse 3-10 photomicrographs so as to count a minimum of 500 BrdU+ cells.
- For data analysis, to assess the fate of BrdU-labeled retinal progenitor cells, we analyse a minimum of 3 cryosections from each pellet (Figures 4A and 4B). We take photomicrographs of each immunostained cryosection, and count the total number of BrdU+ cells and the number of BrdU+ cells that express a cell type-specific marker. For amacrine cells, as an example, we use Pax6 as a cell type-specific marker (Figure 4A). The percentage of BrdU+ progenitor cells that differentiate into Pax6+ amacrine cells is calculated by the formula: %Pax6+BrdU+/BrdU+ cells (as in Tachibana et al., 2016).
- For statistical analysis, we use GraphPad Prism. To compare two data sets, we perform a Student’s t-test. To compare three data sets, we perform a One-way ANOVA and post-hoc Tukey correction (as in Tachibana et al., 2016).
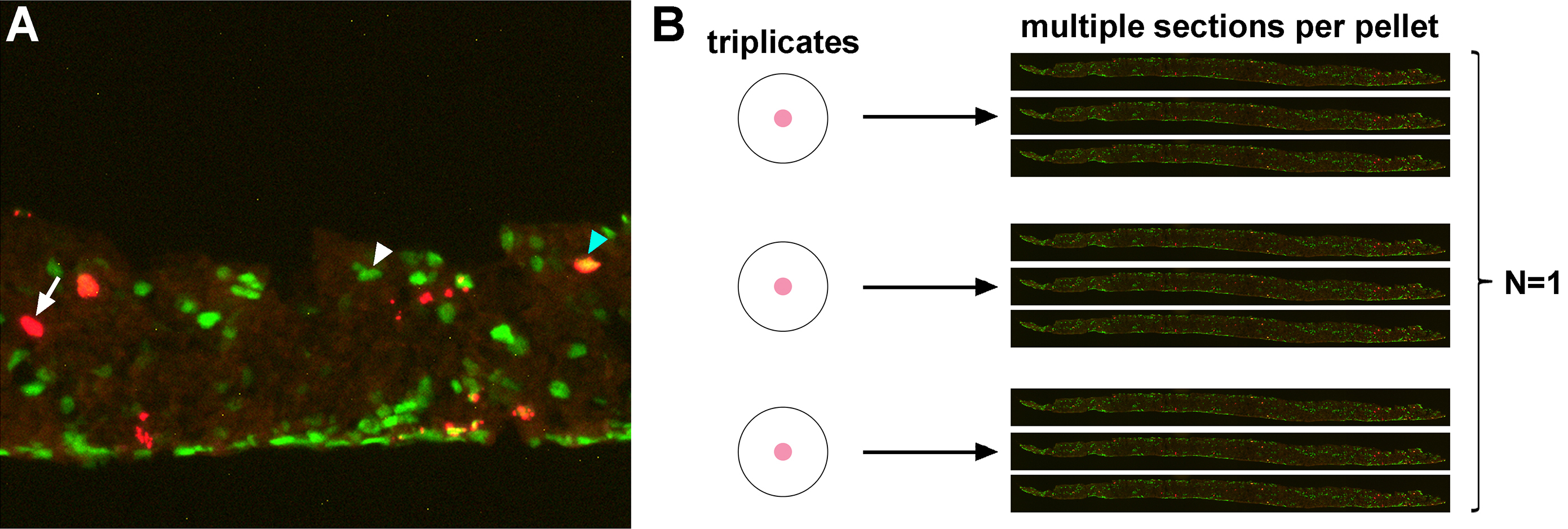
Figure 4. Data processing and analysis of Pax6+BrdU+ cells in the pellet. A. High magnification image of a cryosection of a pellet co-immunostained for Pax6 and BrdU. Arrow/Arrowheads point to BrdU+ cell (arrow; left), Pax6+ cell (white arrowhead; middle) and Pax6+BrdU+ cell (blue arrowhead; right). B. Representative image of how we process data. Number of BrdU+ and Pax6+BrdU+ cells are counted from multiple sections (at least three sections) per pellet, which are at least in triplicate. We combine the data from all sections from each replicate, and consider this as N = 1. To avoid bias, we repeat the experiment at least three times from different litters from different parents.
Notes
- CD1 male and female mice are crossed and embryos are staged considering the morning a vaginal plug is detected as E0.5. E15.5 CD1 embryos, for example, are collected 15 days after
the day of the plug. - Ptenfl/fl; Pax6-Cre+ or Pten+/+ mice are generated by crossing a Ptenfl/+ female with a Ptenfl/+; Pax6-Cre+ male, considering the day the pups are born as P0. P2 embryos are collected 2 days after the day of birth.
- No alterations to the protocol are required if different mouse strains are used.
- When removing RPE and lens from the eyes, it is extremely crucial not to damage the retina as you could lose some cells. While the eye is larger at P2 compared to E15.5, at the macroscopic level there are no visible differences between E15.5 and P2 eyes except size. Thus, the procedure to remove the RPE and lens from the retina remains the same.
- While the retina is easily dissociated with trypsin, other cell types might not be as easily dissociated. In this case, other agents may be tested for dissociation (e.g., collagenase, papain, TrypLETM). Cell strainers can be used to remove clumps as an alternative for tissues that are difficult to dissociate.
- When making the aggregate pellets, we highly recommend to make at least five replicates per sample as you may lose pellets during culturing (e.g., If they sink). This will allow you to have at least three replication controls for each experiment, which should provide a good indication of experimental variability.
- When dislodging the pellet from the bottom of the 15 ml tube, do not apply too much force as it is easy to break apart the pellet. We usually pipet 500 μl of liquid using a P1000 pipetman and slowly dispense liquid on the edge of the pellet. You should see the pellet float in the liquid and then it is easy to pick up.
Recipes
- 2.5% trypsin (40 ml)
- Weigh 1 g of trypsin powder in 50 ml tube and add 40 ml of 1x DPBS
- Vortex until the solution is fully dissolved
- Filter sterilize (0.22 μm), aliquot and store at -80 °C
- Dilute to 0.125% with 1x DPBS for working solution (make fresh prior to use)
- 20% FBS/1x DPBS (10 ml) (make fresh prior to use)
In 8 ml 1x DPBS, add 2 ml FBS
Filter sterilize (0.22 μm) and store at room temperature - Retinal explant media (REM) (50 ml)
Add all of the components:
25 ml DMEM
12.5 ml HBSS (1x)
12.5 ml heat inactivated horse serum
50 μl L-glutamine (200 mM)
300 μl HEPES (1 M)
500 μl penicillin-streptomycin (100x)
100 μl amphotericin B
Filter sterilize (0.22 μm) and store at 4 °C for up to 1 month - 1 mg/ml BrdU (10 ml)
- Weigh 10 mg of BrdU in 15 ml tube and add 10 ml of 1x DPBS
- Vortex until the powder is fully dissolved
- Aliquot and store at -20 °C
- Weigh 10 mg of BrdU in 15 ml tube and add 10 ml of 1x DPBS
- 10x phosphate-buffered saline (PBS) (2 L)
- Mix 163.6 g NaCl, 23 g Na2HPO4, 3.7 g KCl, 4.06 g KH2PO4
- Bring to 2 L with distilled H2O
- Autoclave for 20 min at 121 °C
- 10x PBS is kept at room temperature up to 1 year
- Dilute to 1x with distilled H2O, and adjust pH to 7.5 for working solution
- The 1x PBS can be kept at room temperature for up to 3 months
- Mix 163.6 g NaCl, 23 g Na2HPO4, 3.7 g KCl, 4.06 g KH2PO4
- 20% paraformaldehyde (PFA) (200 ml)
- Weigh 40 g of crystalline PFA in a beaker and add 200 ml of 1x PBS
- Heat the solution until the powder is fully dissolved to 65 °C (not higher as PFA will degrade)
- Aliquot and store at -20 °C
- Dilute to 4% with 1x PBS and adjust pH to 7.5 for working solution (make fresh prior to use)
- Weigh 40 g of crystalline PFA in a beaker and add 200 ml of 1x PBS
- 20% sucrose (500 ml)
- Weigh 100 g of sucrose in a beaker and add 500 ml of 1xPBS
- Heat the solution and mix gently until the powder is fully dissolved
- Filter sterilize (Bottle Top) and store at 4 °C up to 6 months
- Weigh 100 g of sucrose in a beaker and add 500 ml of 1xPBS
- 2 N hydrochloric acid (118 ml) (make fresh)
- Measure 98 ml distilled H2O in a flask and add 20 ml hydrochloric acid gently in the fume hood
- Pre-warm to 37 °C prior to BrdU immunostaining
- Measure 98 ml distilled H2O in a flask and add 20 ml hydrochloric acid gently in the fume hood
- 1x phosphate-buffered saline/0.1% Triton X-100 (PBT) (2 L)
- Dilute 10x PBS (200 ml) to 1x with distilled H2O (1,800 ml)
- Add 2 ml of Triton X-100 and adjust pH to 7.5
- The solution can be kept at room temperature for up to 2 weeks
- Dilute 10x PBS (200 ml) to 1x with distilled H2O (1,800 ml)
- Blocking solution (10% horse serum/1x PBT) (5 ml) (make fresh prior to use)
Aliquot 500 μl of horse serum into 15 ml tube and add 4.5 ml 1x PBT
Vortex to mix thoroughly - Primary antibodies are diluted in blocking solution as described in Table 1
- Secondary antibodies are diluted 1/500 in PBT. DAPI pre-made commercial solution is added at 1/500. Secondary antibodies are listed in Table 1.
Table 1. List of primary and secondary antibodies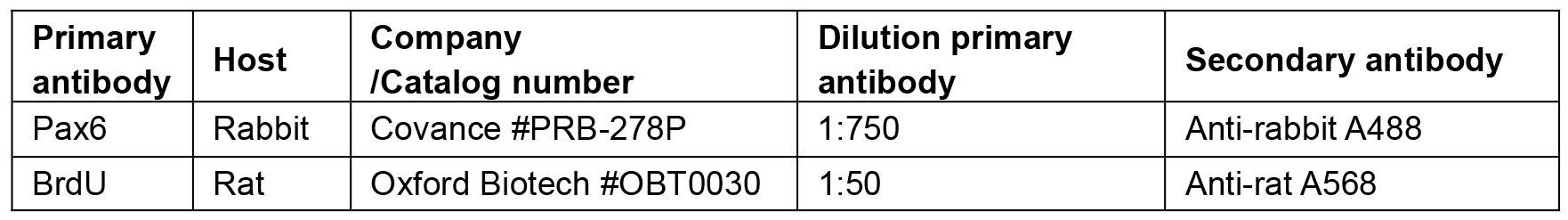
Acknowledgments
This work was supported by grants from Brain Canada to CS and VW and the Canadian Institute of Health Research (CIHR) (Grant #89994) and Lion’s Sight Centre to CS. CS is the Dixon Family Chair in Ophthalmology Research at Sunnybrook Research Institute. NT was supported by an Alberta Children’s Hospital Research Institute (ACHRI)-CIHR scholarship. This protocol was adapted from procedures published in Tachibana et al. (2016) and Ma et al. (2007).
References
- Belliveau, M. J. and Cepko, C. L. (1999). Extrinsic and intrinsic factors control the genesis of amacrine and cone cells in the rat retina. Development 126(3): 555-566.
- Harmon, E. B., Apelqvist, A. A., Smart, N. G., Gu, X., Osborne, D. H. and Kim, S. K. (2004). GDF11 modulates NGN3+ islet progenitor cell number and promotes beta-cell differentiation in pancreas development. Development 131(24): 6163-6174.
- Jensen, A. M. and Wallace, V. A. (1997). Expression of Sonic hedgehog and its putative role as a precursor cell mitogen in the developing mouse retina. Development 124(2): 363-371.
- Lui, J. C. and Baron, J. (2011). Mechanisms limiting body growth in mammals. Endocr Rev 32(3): 422-440.
- Ma, L., Cantrup, R., Varrault, A., Colak, D., Klenin, N., Gotz, M., McFarlane, S., Journot, L. and Schuurmans, C. (2007). Zac1 functions through TGFbetaII to negatively regulate cell number in the developing retina. Neural Dev 2: 11.
- Reh, T. A. and Tully, T. (1986). Regulation of tyrosine hydroxylase-containing amacrine cell number in larval frog retina. Dev Biol 114(2): 463-469.
- Ringuette, R., Wang, Y., Atkins, M., Mears, A. J., Yan, K. and Wallace, V. A. (2014). Combinatorial hedgehog and mitogen signaling promotes the in vitro expansion but not retinal differentiation potential of retinal progenitor cells. Invest Ophthalmol Vis Sci 55(1): 43-54.
- Tachibana, N., Cantrup, R., Dixit, R., Touahri, Y., Kaushik, G., Zinyk, D., Daftarian, N., Biernaskie, J., McFarlane, S. and Schuurmans, C. (2016). Pten regulates retinal amacrine cell number by modulating Akt, Tgfbeta, and Erk signaling. J Neurosci 36(36): 9454-9471.
- Tobin, J. F. and Celeste, A. J. (2005). Myostatin, a negative regulator of muscle mass: implications for muscle degenerative diseases. Curr Opin Pharmacol 5(3): 328-332.
- Wallace, V. A. (2011). Concise review: making a retina--from the building blocks to clinical applications. Stem Cells 29(3): 412-417.
- Waid, D. K. and McLoon, S. C. (1998). Ganglion cells influence the fate of dividing retinal cells in culture. Development 125(6): 1059-1066.
- Wang, Y., Dakubo, G. D., Thurig, S., Mazerolle, C. J. and Wallace, V. A. (2005). Retinal ganglion cell-derived sonic hedgehog locally controls proliferation and the timing of RGC development in the embryonic mouse retina. Development 132(22): 5103-5113.
- Watanabe, T. and Raff, M. C. (1990). Rod photoreceptor development in vitro: intrinsic properties of proliferating neuroepithelial cells change as development proceeds in the rat retina. Neuron 4(3): 461-467.
- Watanabe, T., Voyvodic, J. T., Chan-Ling, T., Sagara, H., Hirosawa, K., Mio, Y., Matsushima, S., Uchimura, H., Nakahara, K. and Raff, M. C. (1997). Differentiation and morphogenesis in pellet cultures of developing rat retinal cells. J Comp Neurol 377(3): 341-350.
- Wu, H. H., Ivkovic, S., Murray, R. C., Jaramillo, S., Lyons, K. M., Johnson, J. E. and Calof, A. L. (2003). Autoregulation of neurogenesis by GDF11. Neuron 37(2): 197-207.
- Zhang, X. M. and Yang, X. J. (2001). Regulation of retinal ganglion cell production by Sonic hedgehog. Development 128(6): 943-957.
Article Information
Copyright
© 2017 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Tachibana, N., Zinyk, D., Ringuette, R., Wallace, V. and Schuurmans, C. (2017). Heterochronic Pellet Assay to Test Cell-cell Communication in the Mouse Retina. Bio-protocol 7(3): e2117. DOI: 10.21769/BioProtoc.2117.
-
Tachibana, N., Cantrup, R., Dixit, R., Touahri, Y., Kaushik, G., Zinyk, D., Daftarian, N., Biernaskie, J., McFarlane, S. and Schuurmans, C. (2016). Pten regulates retinal amacrine cell number by modulating Akt, Tgfbeta, and Erk signaling. J Neurosci 36(36): 9454-9471.
Category
Neuroscience > Cellular mechanisms > Intracellular signalling
Neuroscience > Sensory and motor systems > Retina
Cell Biology > Cell signaling > Intracellular Signaling
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.