- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Experimental Pipeline for SNP and SSR Discovery and Genotyping Analysis of Mango (Mangifera indica L.)

(*contributed equally to this work) Published: Vol 6, Iss 16, Aug 20, 2016 DOI: 10.21769/BioProtoc.1910 Views: 11559
Reviewed by: Samik BhattacharyaGazala AmeenAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Dec 2015
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
Establishing a reservoir of polymorphic markers is an important key for marker-assisted breeding. Many crops are still lack of such genomic infrastructure. Single nucleotide polymorphisms (SNPs) and simple sequence repeats (SSRs) are useful as markers because they are widespread over the genome and many technologies were developed for high throughput genotyping. We present here a pipeline for developing a reservoir of SNP and SSR markers for Mangifera indica L. as an example for fruit tree crops having no genomic information available. Our pipeline includes de novo assembly of reference transcriptome with MIRA and CAP3 based on reads produced by 454-GS FLX technology; Polymorphic loci discovery by alignment of Illumina resequencing to the transcriptome reference; Identifying a subset of loci that are polymorphic in the entire germplasm collection for downstream diversity analysis by genotyping with Fluidigm technology.
Keywords: SNP discoveryBackground
Considerations of high-throughput sequencing: This pipeline does not include RNA/DNA extraction and other molecular biology lab protocols for next generation sequencing (NGS). It is common to outsourcing NGS. Therefore, it includes DNA preparation for genotyping only. Before describing the pipeline below, we would like to comment about the considerations regarding the sequencing.
Assumption: In this pipeline, we assume a non-model organism which has no genomic infrastructure at all. For marker discovery, one will need a reference and resequencing to discover the polymorphism. The ultimate reference is a genome. However, due to the fact that having a good draft or a complete reference genome is still expensive task our recommendation is to sequence a reference transcriptome from a pool of tissues. The pool of tissues should compensate the unequal gene representation as a result of tissue-specific expression.
Technology: For the purpose of a reference transcriptome sequencing, 454-GS Flx Titanium or any long reads NGS technology is preferred. For marker discovery by resequencing, a pool of genomic DNA (gDNA) from the population under study is a cost-effective solution. Polymorphic loci in such pool are a representative sample of the polymorphic loci in the population. Here the important factor is the reads’ depth which should strive to an average coverage of 50x and no less than 20x. In a case of large genomes the choice of gDNA resequencing might be too expensive to get coverage of 50x. Alternatively, mRNA extraction of a pool of tissues and population individuals would be a cheaper option.
The aim of this protocol is to provide a pipeline (Figure 1) for the bioinformatics and genomics support unit that assist the breeder of a crop which has no genomic information to establish a set of polymorphic SNP and SSR markers. This set can be used for marker-assisted breeding studies as well as for exploring the diversity in the crop’s germplasm collection diversity.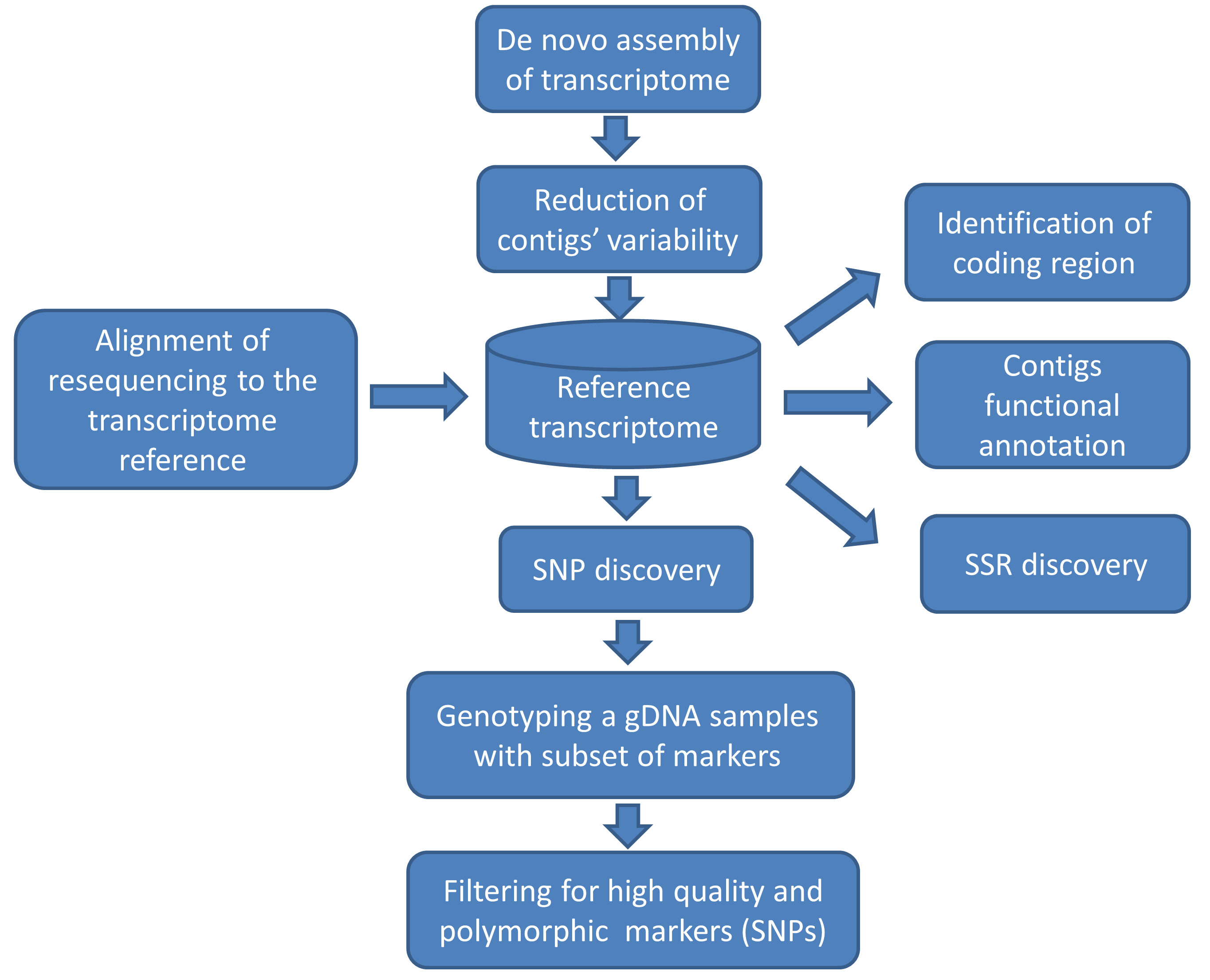
Figure 1. Flowchart of a pipeline for marker discovery. The reference transcriptome here (represented as a database shape) is the link connecting function annotation with genetic variation.
Materials and Reagents
- 50 ml Falcon tube
- Young leaf tissue
- Tris (Amresco, catalog number: 77-86-1 )
- EDTA (Sigma-Aldrich, catalog number: E5134 )
- NaCl (Sigma-Aldrich, catalog number: S3014 )
- 3% CTAB (Hexadecylrimethylammonium bromide) (Sigma-Aldrich, catalog number: H5882 )
- 2% polyvinylpyrolidone (PVP) (MW 40,000) (Amresco, catalog number: 9003-39-8 )
- 1% β-mercaptoethanol (Sigma-Aldrich, catalog number: M3148 )
- 5 M ammonium acetate (Sigma-Aldrich, catalog number: A1542 )
- Chloroform:isoamyl alcohol mix [24:1 (v:v)]
- Isopropanol (stored at -20 °C)
- Ethanol
- RNase A (> 70 Kunit/mg protein, > 20 mg protein/ml) (Sigma-Aldrich, catalog number: R4642 )
- Extraction buffer (see Recipes)
- TE buffer (see Recipes)
Equipment
- 65 °C water bath
- 37 °C water bath/block
- IKA-A11 analytical grinding mill (IKA®-Werke GmbH & Co. KG)
- Cooled centrifuge (Sorvall RC5plus) with Fixed Angle Rotor (FiberliteTM F13-14 x 50cy) (Thermo Fisher Scientific, catalog number: 096-1450 ).
- Agarose gel apparatus
- Nanodrop spectrophotometer
- Recommended hardware specifications (for bioinformatics pipeline)
- CPU
Architecture: x86_64
CPU op-mode(s): 64-bit, 8 cores, Thread(s) per core: 2
Vendor ID: GenuineIntel
CPU MHz: 1596.000 - Memory
MemTotal: 48 GB
SwapTotal: 4GB
Software
- “Sff_extract” (https://bioinf.comav.upv.es/sff_extract/) – Converting and preprocessing, e.g., adapter removal and base-call clipping 454-GS FLX raw files to text formats (fasta and quality).
Note: Sff_extract is now part of the tool set seq_crumbs (https://bioinf.comav.upv.es/seq_crumbs/) - MIRA (https://sourceforge.net/projects/mira-assembler/) – A multi-pass DNA sequence data assembler/mapper for whole genome and/or transcriptome projects. MIRA is a multi-platforms assembler capable assembling reads from a combination of platforms or from each platform separately.
- CAP3 (http://seq.cs.iastate.edu/cap3.html) – CAP3 is for small-scale assembly of sequences with or without quality values.
- Trimmomatic (http://www.usadellab.org/cms/?page=trimmomatic) – Trimmomatic is a fast, multi-threaded command line tool that can be used to trim and crop Illumina (FASTQ) data as well as to remove adapters.
- FASTX (http://hannonlab.cshl.edu/fastx_toolkit/) – Preprocessing, e.g., adapter removal and base-call clipping, short reads (fastq files).
Note: FASTX-Toolkit is a collection of command line tools for Short-Reads FASTA/FASTQ files preprocessing. - Bowtie2 (http://bowtie-bio.sourceforge.net/bowtie2/index.shtml) – Alignment of short reads to a reference genome/transcriptome.
- Samtools (http://www.htslib.org/) – SAM (Sequence Alignment/Map) format is a generic format for storing large nucleotide sequence alignments. SAMTools provide various utilities for manipulating alignments in the SAM format, including sorting, merging, indexing and generating alignments in a per-position format.
- Getorf (http://emboss.sourceforge.net/download/) – EMBOSS tool for identification of open reading frame ORF in mRNA sequence.
- MIcroSAtellite (MISA) identification tool (http://pgrc.ipk-gatersleben.de/misa) – This tool allows the identification and localization of perfect microsatellites as well as compound microsatellites which are interrupted by a certain number of bases.
- SciRoKo (http://kofler.or.at/bioinformatics/SciRoKo/index.html) – A tool for fast whole-genome microsatellite search. For example, the whole rice genome may be searched in 55 sec.
- VarScan (http://varscan.sourceforge.net/) – VarScan is a platform-independent mutation caller for targeted, exome, and whole-genome resequencing data generated on Illumina, SOLiD, Life/PGM, Roche/454, and similar instruments.
Data analysis
- Data
- 454-GS FLX Titanium mRNA contigs were deposited in transcriptome shotgun assembly (TSA) repository of NCBI: accession No. GBJO00000000 (Sherman et al., 2015).
- Illumina short reads were deposited in short reads archive (SRA) of NCBI: experiment accession No. SRX651793 GBJO00000000 (Sherman et al., 2015).
- 454-GS FLX Titanium mRNA contigs were deposited in transcriptome shotgun assembly (TSA) repository of NCBI: accession No. GBJO00000000 (Sherman et al., 2015).
- De novo transcriptome assembly
- Raw sequence reads of the 454-GS FLX Titanium platform were pre-processed by “Sff_extract” (https://bioinf.comav.upv.es/sff_extract/) and arguments for removing the adaptors and clipping the poly-A were applied.

- De novo assembly with MIRA 3.2 (Chevreux et al., 2004)

- Reduction of contig variability (merging transcript variants) by running Cap3 and creating super-contigs
Note: Cap3 is downloaded separately from MIRA (see Software list section).
Note: mango.fasta and mango.qual are output files of MIRA, created in the mango_d_results directory.
- Filtering out contigs with length less than 200 bp
Refer to the fasta file from here on as reference.transcriptome.contigs.fasta
- Raw sequence reads of the 454-GS FLX Titanium platform were pre-processed by “Sff_extract” (https://bioinf.comav.upv.es/sff_extract/) and arguments for removing the adaptors and clipping the poly-A were applied.
- Functional annotation
- Identifying the coding region to annotate marker position, i.e., inside or outside coding sequence. Finding open reading frames (ORFs) by the “getorf” program of the EMBOSS package (Rice et al., 2000). The longest ORF with start and stop codons was chosen for each contig (-find 1) with a minimum cutoff of 50 amino acids (-minisize 150).
Note: The argument (-minimize) is given in base pairs (50 bp x 3 = 150 bp).
- Reference transcriptome contigs annotation to connect variability with functionality using Blast 2GO (Gotz et al., 2008).
Blast2GO GUI options:- Start → load sequences (e.g., fasta)
- Blast → Run Blast Description Annotator
- Mapping → Run mapping
- Annot → Run annotation
- InterPro → Run interproscan

- Start → load sequences (e.g., fasta)
- Identifying the coding region to annotate marker position, i.e., inside or outside coding sequence. Finding open reading frames (ORFs) by the “getorf” program of the EMBOSS package (Rice et al., 2000). The longest ORF with start and stop codons was chosen for each contig (-find 1) with a minimum cutoff of 50 amino acids (-minisize 150).
- SNP and SSR discovery
Adapter removal and low-quality base pairs clipping are performed by Trimmomatic (Bolger et al., 2014) and FASTX (http://hannonlab.cshl.edu/fastx_toolkit/).
Note: Optional but highly recommended if the alignment is performed on RNA-Seq.
- Use R1 and R2 of trimmed pair files, i.e., sample_name_pair_L001_R1.fastq, sample_name_pair_L001_R2.fastq for downstream analysis.
- Alignment of resequencing of Illumina HiSeq-2000 reads to the transcriptome reference with bowtie2 (http://bowtie-bio.sourceforge.net/bowtie2/index.shtml).

- Running samtools (http://www.htslib.org/) and VarScan (Koboldt et al., 2009) for SNP discovery
Note: The criteria for selecting an SNP subset are dependent on the project. However, a few criteria are advisable to ensure confident SNP loci in any project:- No other SNPs in the flanking regions (100 bp each side) to enable of primer design for further analyses (Absolute differences of the SNP value between the previous and next values in ‘Position’ column should > 100).
- Only one SNP per reference-transcriptome contig (unique value at ‘Chrom’ column).
- Bi-allelic confidence (‘SamplesHet’ value > 0).

- No other SNPs in the flanking regions (100 bp each side) to enable of primer design for further analyses (Absolute differences of the SNP value between the previous and next values in ‘Position’ column should > 100).
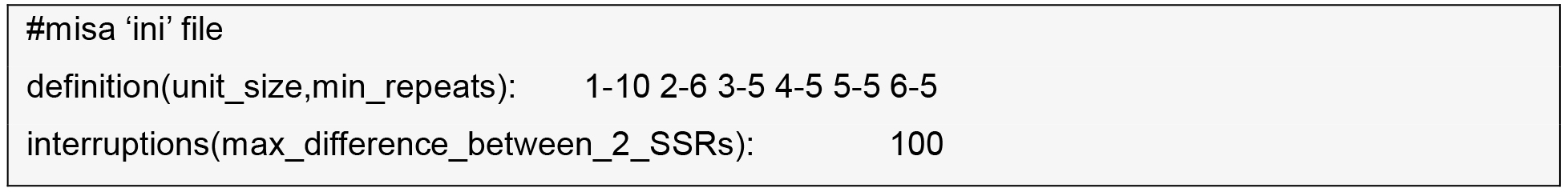
- SSR discovery within the contigs the reference transcriptome.
MIcroSAtellite (MISA) identification tool (http://pgrc.ipk-gatersleben.de/misa) and SciRoKo (Kofler et al., 2007) are run with default parameters.

- Find the intersection between two tables by importing them into MS-Access or SQLite and run a SQL inner join command on contig-name, motif, and start-position.

- Genotyping with Fluidigm
- Large-scale genomic DNA extraction for sample genotyping isolated from young leaves
- Young developing Mango (Mangifera indica L.) leaves were collected from the orchard, frozen in liquid nitrogen and stored at -80 °C until used.
- β-mercaptoethanol was added to extraction buffer which was pre-heated to 65 °C in a pre-warmed water bath.
- 2 g of young leaf tissue was ground to a fine powder using IKA-A11 analytical grinding mill or with a mortar and pestle.
- Ground tissue was transferred to a 50 ml Falcon tube and extracted with 15 ml pre-warmed extraction buffer. Extraction was performed by incubation for 30 min at 65 °C, with occasional mixing of the tube.
- 15 ml of chloroform:isoamyl alcohol mix (24:1, v:v) was added to tubes. Samples were mixed and centrifuged at 17,000 x g for 10 min at 4 °C.
- The aqueous phase was transferred to a new 50 ml tube, and re-extracted with 15 ml of chloroform:isoamyl alcohol mix (24:1, v:v). Centrifugation was performed as above.
- The aqueous phase was transferred to a new tube. 1 volume of ice-cold isopropanol was added, tubes were mixed and DNA was precipitated by centrifugation at 17,000 x g for 20 min at 4 °C.
- Supernatant was carefully disposed of. Pellet was washed with 70% ice-cold ethanol, and centrifuged at 17,000 x g for 10 min at 4 °C.
- Supernatant was carefully disposed of. Pellet was left to dry at room temperature until it turns translucent), and suspended in 3 ml of TE buffer.
- DNA solution was treated with 3 μl RNase A, and incubated for 30 min in 37 °C.
- DNA is precipitated by adding 1/10 volume of 5 M ammonium acetate, and 2/5 volumes of cold 100% ethanol. Tubes are mixed and centrifuged at 17,000 x g for 10 min at 4 °C.
- The supernatant was carefully disposed of. Pellet was washed with 70% ice-cold ethanol, and centrifuged at 17,000 x g for 10 min at 4 °C.
- Pellet is air dried and final DNA is suspended in 200 μl of double distilled water or TE buffer.
- DNA concentration and quality is analyzed on a 0.7% TAE agarose gel and with a Nanodrop spectrophotometer.
- Genotyping on Fluidigm – EP1 Fluidigm standard protocols for FR96.96 chip with four no-template controls (NTCs) instead of one.
Briefly, the protocol is divided into two major sub-protocols – pre-amplification and the assay itself:- First, specific target amplification (STA) protocol is performed to have an approximately equal proportion from each target by running the following steps:
1) Preparing the 10x SNPtype STA. Primer pool for 96 assays.
2) Performing STA on a PCR machine.
3) Dilution of samples (the outcome will be used in stage 4 of the second part). - Second, the assay of genotyping by specific target primers is performed in a Fluidigm 96.96 dynamic genotyping array on the EP1 platform as follow:
1) Priming the 96.96 Dynamic ArrayTM IFC.
2) Preparing SNPtype assays mixes.
3) Preparing 10x Assays.
4) Preparing Sample Pre-Mix and Samples.
5) Loading the Chip.
6) Using the FC1TM Cycler.
7) Using the EP1TM Reader Data Collection Software.
8) Extracting the data for downstream bioinformatics analysis.
- First, specific target amplification (STA) protocol is performed to have an approximately equal proportion from each target by running the following steps:
- Filtering qualified SNPs for diversity analysis
Fluidigm genotype calls are divided into four categories by visual inspection:- Filtering out SNPs with a Category ≥ 2 (Table 1).
- Filtering out SNPs with more than 10% no calls.
- Filtering out samples with more than 33% no calls.
- Filtering out markers with PIC < 0.1.
- Filtering out markers with more than 90% of the samples have the same call, i.e., segregating exactly the same.
- Filtering out markers with less than 2 samples in each genotype.
- Leaving only one marker from each pair of linked markers (R^2 > 0.7).
- Leaving only one sample from group of sample having identical genotype. (Identity ≥ 0.95)
*Notes:- These steps can be performed with any programming language, e.g., R, python, perl, C, etc. or SQL.
- PIC is calculated as PIC = 1- ∑ pi^2; i = a, A
- R^2 is calculated as r^2 = D^2/(p1*p2*q1*q2); D = (p11*p22)-(p12p21 p11,p22,p12,p21 are the proportions of all possible combinations of two bi-allelic loci.
Table 1. Quality scores of each locus genotyping calls given by visual inspection.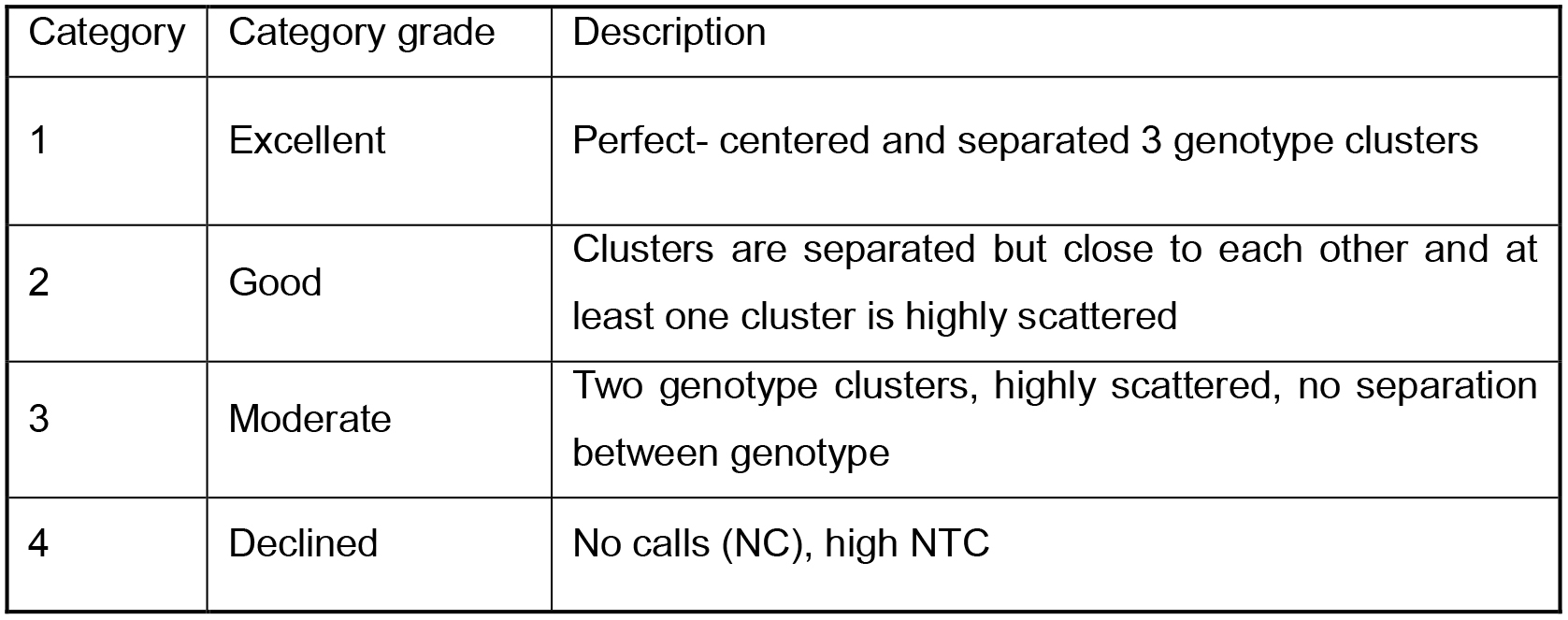
- Filtering out SNPs with a Category ≥ 2 (Table 1).
- Use R1 and R2 of trimmed pair files, i.e., sample_name_pair_L001_R1.fastq, sample_name_pair_L001_R2.fastq for downstream analysis.
Recipes
- Extraction buffer
100 Tris, pH 8
20 M EDTA
1.5 M NaCl
3% hexadecylrimethylammonium bromide (CTAB)
2% polyvinylpyrolidone (PVP)
1% β-mercaptoethanol
All solution except β-mercaptoethanol are dissolved by stirring over several hours, and autoclaved. β-mercaptoethanol is added just prior to tissue extraction. - TE buffer
10 mM Tris, pH 8.0
1 mM EDTA
Acknowledgments
The protocol has been developed in a study which was supported by the Chief Scientist of Ministry of Agriculture and Rural Development [Grant No.: 203-0859-12].
References
- Bolger, A. M., Lohse, M. and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30(15): 2114-2120.
- Bowtie - An ultrafast memory-efficient short read aligner. JOHNS HOPKINS University.
- Chevreux, B., Pfisterer, T., Drescher, B., Driesel, A. J., Muller, W. E., Wetter, T. and Suhai, S. (2004). Using the miraEST assembler for reliable and automated mRNA transcript assembly and SNP detection in sequenced ESTs. Genome Res 14(6): 1147-1159.
- Fluidigm SNP Genotyping Guide. Fluidigm.
- FASTX-Toolkit. Hannonlab.
- Gotz, S., Garcia-Gomez, J. M., Terol, J., Williams, T. D., Nagaraj, S. H., Nueda, M. J., Robles, M., Talon, M., Dopazo, J. and Conesa, A. (2008). High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res 36(10): 3420-3435.
- Koboldt, D. C., Chen, K., Wylie, T., Larson, D. E., McLellan, M. D., Mardis, E. R., Weinstock, G. M., Wilson, R. K. and Ding, L. (2009). VarScan: variant detection in massively parallel sequencing of individual and pooled samples. Bioinformatics 25(17): 2283-2285.
- Kofler, R., Schlotterer, C. and Lelley, T. (2007). SciRoKo: a new tool for whole genome microsatellite search and investigation. Bioinformatics 23(13): 1683-1685.
- MIcroSAtellite identification tool.
- Reading/writing/editing/indexing/viewing SAM/BAM/CRAM format. Samtools.
- Rice, P., Longden, I. and Bleasby, A. (2000). EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet 16(6): 276-277.
- Sff_extract. Bioinformatics at COMAV.
- Sherman, A., Rubinstein, M., Eshed, R., Benita, M., Ish-Shalom, M., Sharabi-Schwager, M., Rozen, A., Saada, D., Cohen, Y. and Ophir, R. (2015). Mango (Mangifera indica L.) germplasm diversity based on single nucleotide polymorphisms derived from the transcriptome. BMC Plant Biol 15: 277.
Article Information
Copyright
© 2016 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Sharabi-Schwager, M., Rubinstein, M., Ish shalom, M., Eshed, R., Rozen, A., Sherman, A., Cohen, Y. and Ophir, R. (2016). Experimental Pipeline for SNP and SSR Discovery and Genotyping Analysis of Mango (Mangifera indica L.). Bio-protocol 6(16): e1910. DOI: 10.21769/BioProtoc.1910.
Category
Plant Science > Plant molecular biology > DNA > Genotyping
Plant Science > Plant molecular biology > DNA > DNA extraction
Molecular Biology > DNA > DNA extraction
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.