- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Micro-chromatin Immunoprecipitation (μChIP) Protocol for Real-time PCR Analysis of a Limited Amount of Cells


Published: Vol 6, Iss 12, Jun 20, 2016 DOI: 10.21769/BioProtoc.1846 Views: 9691
Reviewed by: Salma HasanAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Jul 2015
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
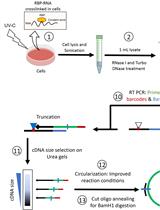
Chromatin immunoprecipitation followed by deep sequencing (ChIP-Seq) is an important strategy to study gene regulation. When availability of cells is limited, however, it can be useful to focus on specific genes to investigate in depth the role of transcription factors or histone marks. Unfortunately, performing ChIP experiments to study transcription factors’ binding to DNA can be difficult when biological material is restricted. This protocol describes a robust method to perform μChIP for over-expressed or endogenous transcription factors using ~100,000 cells per ChIP experiment (Masserdotti et al., 2015). We also describe optimization steps, which we think are critical for this protocol to work and which can be used to further reduce the number of cells.
Keywords: ChromatinMaterials and Reagents
- Microcentrifuge tubes (siliconized polypropylene, 1.7 ml) (Sigma-Aldrich, catalog number: T3406-250EA )
- Dynabeads® Protein G (Thermo Fisher Scientific, NovexTM, catalog number: 10004D )
- SYBR® green master mix (choose according to the Real-time PCR system used)
- Protease cocktail inhibitor (Roche Diagnostics, catalog number: 04693116001 )
- 37% formaldehyde solution (Sigma-Aldrich, catalog number: F8775 )
- Di(N-succinimidyl) glutarate (Sigma-Aldrich, catalog number: 80424 )
- Glycine (Thermo Fisher Scientific, Fisher ScientificTM, catalog number: 10070150 )
Note: 20x glycine stock solution (2.5 M) in water - DNase-free RNase A (ThermoFisher Scientific, catalog number: EN0531 )
- Proteinase K (Thermo Fisher Scientific, AmbionTM, catalog number: AM2546 )
- 8 M lithium chloride solution (LiCl) (Sigma-Aldrich, catalog number: L7026 )
- DynaMag®-2 magnet (Thermo Fisher Scientific, NovexTM, catalog number: 12321D )
- DNA MinElute kit (QIAGEN, catalog number: 28004 )
- Histone mark H3K9Acetyl antibody (Abcam plc, catalog number: ab4441 )
- HA antibody (Abcam plc, catalog number: ab9110 )
- SDS (Sigma-Aldrich, catalog number: L3771 )
- EDTA (Sigma-Aldrich, catalog number: 798681 )
- Tris (TRIZMA® base) (Sigma-Aldrich, catalog number: T1503 )
- Bovine serum albumin (BSA) (Sigma-Aldrich, catalog number: A3608 )
Note: 250 mg/ml BSA stock solution in water - HEPES (Sigma-Aldrich, catalog number: H3375 )
- NP-40 (Ipegal®) (Sigma-Aldrich, catalog number: I-3021 )
- Na-DOC (Sigma-Aldrich, catalog number: D6750 )
- NaHCO3 (Sigma-Aldrich, catalog number: S5761 )
- ChIP IP buffer (see Recipes)
- Lysis buffer (see Recipes)
- Lithium chloride (LiCl) washing buffer (see Recipes)
- Tris-EDTA (TE) buffer (see Recipes)
- Elution buffer (see Recipes)
Equipment
- Rotating wheel
- Dry heat block
- Thermomixer (Eppendorf AG)
- Nanodrop
- Water bath sonicator (Diagenode, Bioruptor®)
- Real-time PCR instrument
Procedure
- Optimization steps
These 4 steps are critical and preparatory work is required for this protocol to work optimally.- The chromatin concentration per 100,000 cells has to be estimated for each cell line or tissue used. Follow the protocol up to step C11 and continue from step C28 to reverse the cross-linking, treat with RNase A and proteinase K before purifying the sample through a column. Measure the concentration of the DNA with a nanodrop.
Note: Typical concentration is between 4-6 ng/µl with NS-5 cells giving a yield of chromatin solution between 400-600 ng per 100 μl. This information will be used to optimize the antibody concentration. - The concentration of antibody to be used per 100,000 cells needs to be optimized by initially testing at least 3 different antibody concentrations. If the antibody is commercial, this can be done by working out the optimal concentration per μg of chromatin given on the manufacturer’s datasheet and the estimated chromatin concentration from the previous step. Otherwise, the antibody concentration needs to be tested empirically until an efficient immunoprecipitation is achieved.
We have noticed a maximum antibody concentration for which the immunoprecipitation peaks (see Figures 1-2). - Sonication time needs to be optimized depending on the machine used. As a guideline, in an ice-cold water bath sonicator, we sonicate a volume of 400-500 μl for maximum 10 min (30 sec on/30 sec off on maximum power) keeping a constant layer of ice. Optimal sonicated chromatin is depicted in Figure 3.
- Design primers with Primer3 (http://primer3.ut.ee) and test their specificity by serial dilution of genomic DNA. The amplicon size should be between 100 and 150 bp.
- The chromatin concentration per 100,000 cells has to be estimated for each cell line or tissue used. Follow the protocol up to step C11 and continue from step C28 to reverse the cross-linking, treat with RNase A and proteinase K before purifying the sample through a column. Measure the concentration of the DNA with a nanodrop.
- Before starting
- Plate or sort 100,000 cells by flow cytometry per µChIP including mock ChIP with additional cells for input.
Note: Input can be made with 100,000 cells (100%) or with a fraction of cells bearing in mind the percentage for final quantification. - Resuspend the Dynabeads® and take a sufficient volume for the pre-clearing step and the IP step. Capture on magnet, aspire supernatant and wash with 1 ml ice-cold ChIP IP buffer on a rotating wheel for 3 min at room temperature. Repeat 2 times and resuspend in ChIP IP buffer with the same volume initially pipetted out from stock. Put the tube on a rotating wheel at 4 °C while proceeding with the next steps.
- Plate or sort 100,000 cells by flow cytometry per µChIP including mock ChIP with additional cells for input.
- Experimental procedure
For plated cells, take off media and wash cells with 1x PBS at room temperature. Cross-link the cells directly on plate with sufficient volume of cross-linking solution to cover cells. For sorted cells, use siliconized tubes during sorting, pellet the cells (100 x g for 3 min), wash with 500 ml PBS and cross-link in 1 ml PBS on a rotating wheel (12 rpm).- (Optional) If double cross-linking is required, cross-link first with 2 mM Di(N-succinimidyl) glutarate in PBS for 45 min at room temperature with gentle rocking, then wash twice with PBS.
- Cross-link with 1% formaldehyde in PBS for 10 min with gentle rocking.
- Quench fixation with 125 mM glycine and keep on rocker for 5-10 min.
- Wash twice with cold PBS, on the last wash remove as much PBS as possible with a pipette.
- Lyse cells directly on plate with 100 µl lysis buffer per 100,000 cells (i.e., minimum 300 µl to get one input, one mock and one ChIP sample; 400 µl if 2 different proteins are immunoprecipitated).
- Collect cells with a cell scraper and transfer into a siliconized tube.
Optional: Snap freeze in liquid nitrogen (this step is not required for some antibodies, such as antibodies for histone marks). - Leave to thaw on ice.
- Sonicate in a water bath sonicator with ice for 10 min maximum (see step A3).
- Centrifuge at 4 °C at 16,000 x g for 5 min and transfer supernatant into a new siliconized tube.
- Take an aliquot (~10 µl) and run it on a 1% agarose gel to check shearing (see Figure 3 for optimal result).
Note: Make sure the chromatin runs between 500-1,000 bp before continuing or sonicate longer. - Pre-clear with 10 µl washed Dynabeads® per 100 µl of chromatin solution for 1 h on rotating wheel at 4 °C.
- Capture beads on magnet and collect supernatant into a fresh siliconized tube.
- Aliquot 100 µl in siliconized tubes for the input, the mock ChIP and each ChIP sample(s).
- Add 400 µl ChIP IP buffer to the mock and ChIP sample(s) (SDS concentration has to be reduced to 0.1%).
Note: Keep the input sample(s) at -20 °C until the next day. - Add correct amount of antibody to the diluted mock and ChIP samples and rotate overnight on a wheel at 4 °C.
Note: Keep the remaining of the washed beads on a wheel overnight to fully saturate them with the BSA contained in the buffer. - Next morning, add 25 µl of saturated Dynabeads® and put back on a wheel at 4 °C for 2 h.
- Capture the Dynabeads® on a magnet, take out the supernatant and add 1 ml of ice-cold LiCl washing buffer. Put on a wheel at room temperature for 3-5 min and repeat 4 times.
- Wash 1 time with 1 ml ice-cold TE buffer. Centrifuge briefly before putting on a magnet to remove as much as possible of the TE buffer.
- Elute antibody/chromatin complexes by adding 50 µl of elution buffer directly to washed Dynabeads®.
- Vortex briefly.
- Shake on Thermomixer at 1,000 rpm for at least 15 min at room temperature.
- Centrifuge at maximum speed in a top table centrifuge for 3 min.
- Put tubes on magnet and collect supernatants into fresh siliconized tubes.
- Repeat the elution step and combine elutes in the same tubes.
- Thaw input sample and follow next steps for all samples.
- Add 10 µl 5 M NaCl (final concentration 0.45 M).
- Boil samples in dry heat block for 15 min to reverse crosslinking.
- Spin down tubes at maximum speed for 30 sec at room temperature.
- Leave the tubes to cool down before adding 1 µl DNase-free RNase A.
- Incubate for 15 min at 37 °C.
- Add 0.5 µl proteinase K.
- Incubate for 15 min at 67 °C.
- Purify DNA with a kit designed for low DNA quantity (e.g., DNA MinElute kit).
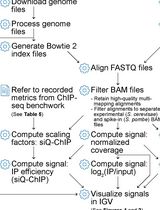
- Quantities of immunoprecipitated DNA are calculated by comparison with a standard curve generated by serial dilutions of input DNA using a Real-Time PCR System and an SYBR green-based kit for quantitative PCR (Frank et al., 2001). As a guideline, we use 1-2 μl of purified DNA per reaction depending on the targeted protein and the number of genes to analyze. The final concentration of precipitated DNA varies according to the targeted protein. In NS-5 cells, we obtained around 4 ng/μl for endogenous transcription factors and up to 30 ng/μl for histone marks.
- (Optional) If double cross-linking is required, cross-link first with 2 mM Di(N-succinimidyl) glutarate in PBS for 45 min at room temperature with gentle rocking, then wash twice with PBS.
Representative data
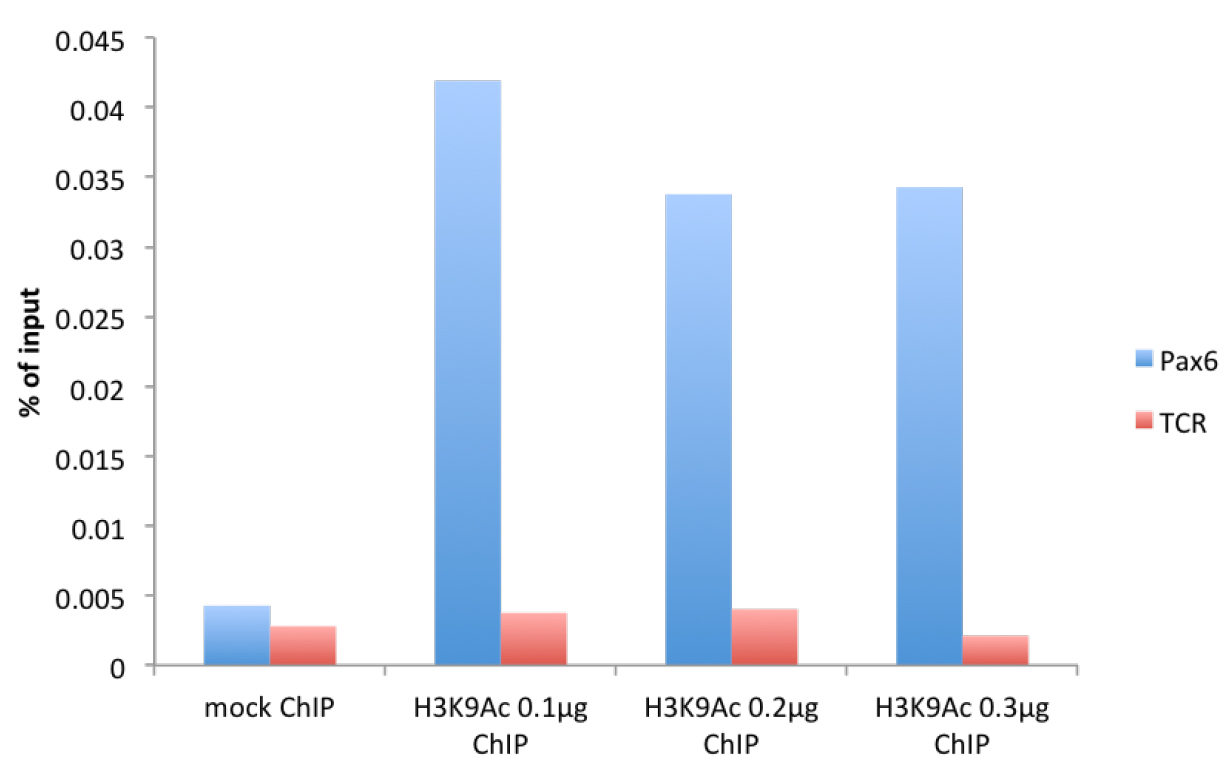
Figure 1. μChIP of the histone mark H3K9Acetyl with increasing amount of antibody in mouse cortical astrocytes. Real-time PCR was performed in the promoter region of Pax6 (F: cggtgaaagaagccactagg, R: agggaacacaccaactttcg) and in the promoter region of T-cell receptor (TCR) (F: cttacaccccaaacctccaa, R: gggaggatgaggagaaaagg).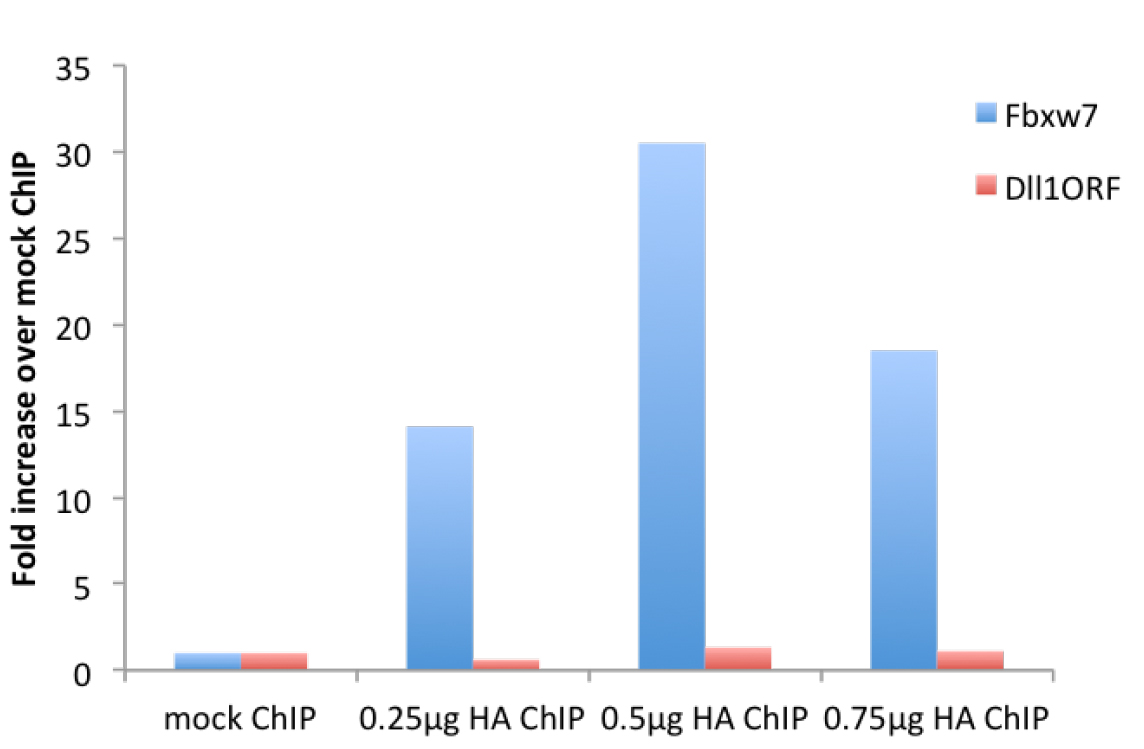
Figure 2. μChIP of overexpressed HA-tagged Neurogenin2 in NS-5 cells with increasing amount of antibody. Real-time PCR was performed with primers recognizing the promoter region of the Neurogenin2 (Neurog2) target gene Fbxw7 (F: gcgctttgaagaaagacctg, R: gttttcaaggggcgaatgta) and the ORF region of Dll1, not bound by Neurog2 (F: gtctcaggaccttcacagtag, R: gagcaaccttctccgtagtag).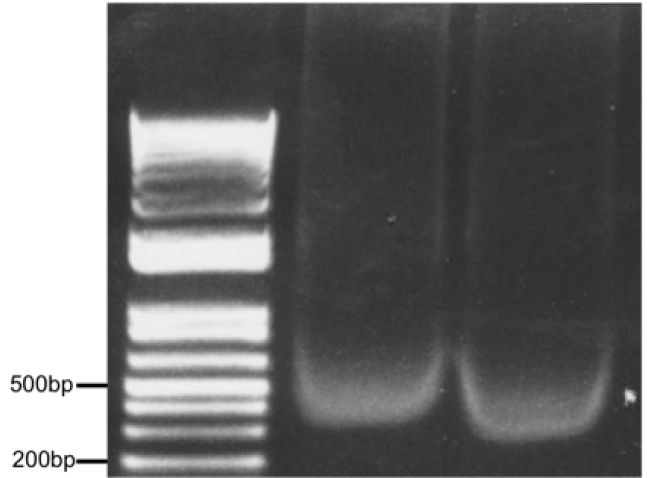
Figure 3. Optimal chromatin solution after sonication of 500,000 astrocytes in 500 μl lysis buffer for 10 min (30 sec on/30 sec off) setting H, Diagenode Bioruptor. Two samples (10 μl each) were run on a 1% agarose gel alongside a DNA ladder.
Recipes
Note: Prepare all the solutions fresh from stock solutions before starting and keep on ice.
- ChIP IP buffer
20 mM HEPES (pH 7.6) from 1 M stock solution
0.2 M NaCl from 5 M stock solution
2 mM EDTA (pH 8.0) from 0.5 M stock solution
0.1% Na-DOC from 10% fresh solution
1% Triton X-100 from 10% fresh solution
5 mg/ml BSA from 250 mg/ml stock solution
Supplemented with protease inhibitor cocktail - Lysis buffer
0.5% SDS from 10% stock solution
10 mM EDTA (pH 8.0) from 0.5 M stock solution
50 mM Tris-HCl (pH 8.0) from 1 M stock solution
Supplemented with 1x protease inhibitor cocktail - Lithium chloride (LiCl) washing buffer
50 mM HEPES (pH 7.6) from 1 M stock solution
1 mM EDTA (pH 8.0) from 0.5 M stock solution
1% NP-40 from 10% fresh solution
1% Na-DOC from 10% fresh solution
0.5 M LiCl from 8 M stock solution - Tris-EDTA (TE) buffer
10 mM Tris-HCl (pH 8.0) from 1 M stock solution
1 mM EDTA (pH 8.0) from 0.5 M stock solution - Elution buffer
50 mM NaHCO3 (pH 5.2) from 3 M stock solution
1% SDS from 10% stock solution
Acknowledgments
This work was supported by a project grant from the Wellcome Trust (WT091800MA) and a Grant-in-Aid from the Medical Research Council (U117570528) to F. G. This protocol was established with methods from Peggy Farnham's laboratory and with methods published by François Guillemot's laboratory. We are grateful to Dr Koji Oishi for critical reading of the manuscript.
References
- Frank, S. R., Schroeder, M., Fernandez, P., Taubert, S. and Amati, B. (2001). Binding of c-Myc to chromatin mediates mitogen-induced acetylation of histone H4 and gene activation. Genes Dev 15(16): 2069-2082.
- Masserdotti, G., Gillotin, S., Sutor, B., Drechsel, D., Irmler, M., Jorgensen, H. F., Sass, S., Theis, F. J., Beckers, J., Berninger, B., Guillemot, F. and Gotz, M. (2015). Transcriptional mechanisms of proneural factors and REST in regulating neuronal reprogramming of astrocytes. Cell Stem Cell 17(1): 74-88.
Article Information
Copyright
© 2016 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Gillotin, S. and Guillemot, F. (2016). Micro-chromatin Immunoprecipitation (μChIP) Protocol for Real-time PCR Analysis of a Limited Amount of Cells. Bio-protocol 6(12): e1846. DOI: 10.21769/BioProtoc.1846.
Category
Molecular Biology > DNA > DNA-protein interaction
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.